Cardiogenic Shock: Emergency Department-Focused Management
- Jan 23rd, 2023
- Mary Hamblen
- categories:
Authors: Mary Hamblen, DO (EM Resident Physician, Christus Spohn/Texas A&M University School of Medicine, Corpus Christi, TX); J.D. Cambron, DO (EM Attending Physician, Christus Spohn/Texas A&M University School of Medicine, Corpus Christi, TX) // Reviewed by: Jessica Pelletier, DO (EM Education Fellow, Washington University School of Medicine, St. Louis, MO); Marina Boushra, MD (EM-CCM Attending Physician, Cleveland Clinic Foundation); Brit Long, MD (@long_brit)
Case
A 56-year-old male with a history of coronary artery disease, alcohol use disorder, and recent myocardial infarction (MI) presents to the emergency department (ED) complaining of shortness of breath for the last day. One month prior, he had a stent placed in his left anterior descending artery (LAD) for 100% occlusion. His hospital stay was complicated by multiple ventricular fibrillation (VF) arrests, and he had an Automatic Implantable Cardioverter Defibrillator (AICD) placed. Today, he is pale, cool, and diaphoretic. His work of breathing is labored, and he has fine crackles in all lung fields on auscultation. His vital signs include a heart rate (HR) of 100 beats per minute (bpm), blood pressure (BP) of 160/100 mm Hg, oxygen saturation of 85% on continuous positive airway pressure (CPAP), respiratory rate (RR) of 30-40 breaths/min, T 97 F°, and a Glasgow Coma Scale (GCS) of 15. In the first five minutes of the presentation, his AICD fires twice in response to two separate episodes of sustained ventricular tachycardia (VT). Successful cardioversion to a narrow complex tachycardia is accomplished that vacillates between atrial fibrillation and atrial flutter at a rate of 100-150.
Introduction
Cardiogenic shock (CS) is the result of pump failure, and it is characterized by the inability of the cardiac output to adequately meet the oxygen needs of the peripheral tissues.1-2 The most common cause is MI, followed by dysrhythmias, heart failure, and valvular disease.3 The downstream cascade has a common pathway regardless of the inciting event: decreased cardiac output leads to myocardial ischemia, which triggers compensatory hormonal responses that paradoxically create a positive feedback loop, promoting further ischemia.3 Although there has yet to be universally accepted criteria to define CS, some of the landmark trials such as SHOCK and IABP-SHOCK propose a cluster of similar descriptors: hypotension, end-organ hypoperfusion, a cardiac index (CI) <2.2 L/min/m2, and high pulmonary capillary wedge pressure (PCWP) ≥15 mmHg (Figure 1).4,5 Not all findings need to be present for the diagnosis of CS to be made.3
Figure 1. Characteristics of cardiogenic shock.

In the ED setting, all four criteria can be evaluated with a good history and physical exam, blood tests, electrocardiogram (ECG), and bedside ultrasound.3 Hypotension in the SHOCK trial was defined as systolic BP (SBP) <90 mmHg, mean arterial pressure (MAP) >30 mmHg below baseline, or need for vasopressors to maintain SBP >90 mmHg.4 End-organ hypoperfusion may be identified by physical exam findings such as altered mentation, decreased urination, or cold extremities, or by lab markers such as serum lactate >2.0 mmol/l.3 Non-invasive CI estimations via point-of-care ultrasound (POCUS) measurement of LV outflow tract (LVOT) velocity time integral (VTI) and LVOT diameter have been repeatedly validated.6,7 Pulmonary congestion, as indicated by rales and low oxygen saturation, is suggestive of high PCWP, but this finding may not always be present.3 Jugular venous pressure (JVP) has been described as another surrogate marker of PCWP, with the ESCAPE trial showing a correlation between JVP ≥12 mm Hg and PCWP ≥22 mm Hg.8
In practice, CS has a variable initial presentation depending on the patient’s degree of decompensation and which side of the heart is most affected.9 There are four classic clinical presentations of CS, which are identified by hemodynamic state and physical examination and correlate with mortality (Table 1). In this classification, the patients are divided into “warm” and “cold” phenotypes, with “warm” referring to the temperature of the extremities, which corresponds to decreased systemic vascular resistance/distal vasodilatation, and “cold” to widespread vasoconstriction. Warm extremities may indicate the presence of systemic inflammatory response syndrome (SIRS) (which is common as CS worsens) or a mixed pattern of shock.9 Patients are further classified by “dry” versus “wet” phenotypes, referring to the condition of the lungs and the presence of pulmonary edema.9 “Wet” almost always indicates left ventricular (LV) failure and high filling pressures which have led to congestion in the lungs.9 Relative mortality in the context of MI is highest at 51% with the “Cold-Wet” type, lowest with the “Warm-Dry” at 3%, and moderate for “Warm-Wet” (9%) and “Cold-Dry” (23%).10
Table 1. Classification of CS by hemodynamic state and physical examination. “Warm” indicates warm extremity temperatures, indicative of distal vasodilatation and low SVR. “Cold” refers to distal vasoconstriction and high SVR. “Wet” indicates pulmonary edema and “dry” the lack thereof. Warm-Dry connotes the best prognosis. Cold-Wet connotes a mortality of 51% from MI. Adapted from Chioncel et al. (2020).9

The first step in treating CS is recognizing it.11 A good first step in evaluating for undifferentiated shock in the emergency department is the Rapid Ultrasound for Shock and Hypotension (RUSH) exam, which has been covered by Daly et al. (2020) in a previous emDocs post (http://www.emdocs.net/diagnosing-cardiogenic-shock-in-the-ed/).11 The RUSH exam is a quick evaluation of some of the key anatomical structures involved in the causes of, and compensation for, different types of shock. The HIMAP mnemonic helps guide the clinician through all parts of the exam: heart, inferior vena cava (IVC), Morrison’s pouch/Extended Focused Assessment with Sonography in Trauma (E-FAST), aorta, and pulmonary.12 Findings on ultrasound consistent with CS are: (1) poor LV systolic function; (2) a plump IVC (>2 cm/does not change >50% with inspiration); (3) ≥ 3 B-lines in 2+ areas of the chest; (4) pleural effusion; (5) peritoneal fluid; (6) RV that is > 2/3 the size of the LV.13
Stabilization
As always, clinicians should obtain a full set of vital signs including pulse oximetry, end-tidal capnography if available, electrocardiogram (ECG), and intravenous (IV) access (in the setting of CS, at least two IVs are preferred). Initial treatment should be focused on optimizing airway/breathing/circulation (ABCs) and searching for reversible causes.3
A – Airway // B – Breathing
Non-invasive positive pressure ventilation (NIPPV) should be started in patients without contraindications who present with increased work of breathing, RR >25 breaths/min, or oxygen saturation <90%.14 In general, NIPPV is beneficial in patients with acute pulmonary edema because it increases pressure within the alveoli, helping to mobilize fluid out of the alveoli and back into the vasculature. It also helps recruit alveoli and keep them open.15 Caution is advised as NIPPV may have some unfavorable effects in patients with CS. High intrathoracic pressure will decrease RV preload and increase pulmonary vascular resistance (RV afterload), which can spur worsening hypotension in an already failing right heart.15 Therefore, in the context of extreme hemodynamic instability, NIPPV is often contraindicated.16 NIPPV is similarly contraindicated in patients with altered mentation, a common finding in CS.16 This does not mean, however, that anyone with CS should immediately be intubated. Hongisto et al. (2017) published data showing no difference in mortality in patients with CS who were randomized to either NIPPV or immediate intubation and mechanical ventilation.16 Although endotracheal intubation is a viable option, it does carry some disadvantages in comparison to NIPPV. Those include but are not limited, to hemodynamic compromise during induction and sedation, risk of ventilator-associated pneumonia, the need for continuous sedation, a need for more intensive critical care monitoring, and more specialized training required for application.15-16 For a CS patient who is awake, breathing, following commands, not actively vomiting, without severe facial deformities, and who is not completely preload-dependent, a trial of NIPPV is reasonable.1
Initial CPAP settings should start at a pressure of 5-10 cmH2O, and be titrated upward by 2 cmH2O to goal oxygen saturation >92%.1 Initial recommended bilevel positive airway pressure (BiPAP) settings are inspiratory positive airway pressure (IPAP) of 10 cmH2O and expiratory positive airway pressure (EPAP) of 5 cmH2O, with titration of IPAP by 2-3 cmH2O as needed to correct hypercapnia. Titrate EPAP upward to maintain oxygen saturation >92%. Avoid IPAP >20 cmH2O to minimize gastric insufflation and aspiration events.1
Patients who fail a trial of NIPPV– defined by mental status decline, refractory hypoxia or hypercapnia, or failure to improve within the first 30 minutes to one hour – should be intubated (see Table 2 for medication suggestions). Further, patients who upon initial presentation have altered mental status, extreme hemodynamic instability/collapse, active vomiting, impending respiratory failure, extreme acid-base derangements, severe hypoxemia or hypercapnia, should not have a trial of NIPPV and instead be intubated.1 Caution is recommended during induction, as too high a dose of sedation medication can result in hemodynamic compromise (Table 2).1
Table 2. Recommended induction agents and paralytics when intubating a patient in cardiogenic shock.1 Note: Caution is recommended with induction medications due to the risk of hypotension. Administration should be carefully titrated to the lowest necessary dose to achieve appropriate sedation.


If intubation and mechanical ventilation are required, low tidal volumes (5-7 ml/kg of ideal body weight) are recommended to decrease pulmonary vascular resistance and stress on the RV.17 Positive end-expiratory pressure (PEEP) settings depend on which side of the heart is weakest; for RV failure, start with PEEP of 3-5 cmH2O, and for LV failure, start with PEEP of 5-10 cmH2O.1 In RV-predominant heart failure (Figure 2), moderate to high PEEP will decrease RV cardiac output by decreasing RV venous return, increasing pulmonary vascular resistance and thus RV afterload. The resultant expansion of the RV diameter from the increased end-diastolic volume causes bowing of the interventricular septum into the LV, leading to decreased LV cardiac output.1 Conversely, for LV-predominant heart failure, moderate to high PEEP will decrease LV preload and afterload, causing increased LV cardiac output.1 Fraction of inspired oxygen (FiO2) should be set at 100% and rapidly titrated down to maintain oxygen saturation > 92%.1 Recommendations regarding ventilator mode do not specify one singular best approach for all patients, but they do advise against modes like synchronized intermittent mandatory ventilation (SIMV).1 SIMV may increase myocardial oxygen demand since it will not support patient-initiated breaths.1
There is a clear consensus that every attempt should be made to avoid intubation in the setting of right heart failure.1 If intubation is necessary, awake intubation, facilitated with ketamine, is probably the preferred approach due to the loss of sympathetic drive that occurs during rapid-sequence intubation.1 Systemic blood pressure needs to be kept higher than the pulmonary artery pressure, which means having vasopressors ready or already running at the time of intubation. Norepinephrine is frequently cited as the first-line vasopressor in this situation.21 Avoiding hypoxia and hypercapnea is essential.1 Post-intubation peak airway pressures should be kept to a minimum in order to minimize intrathoracic pressure, since elevated intrathoracic pressure will reduce preload and worsen CS.1
Note: 60-80% of patients with CS will ultimately require invasive mechanical ventilation.9
Figure 2. PEEP settings for RV or LV failure. In general, patients with RV-predominant failure benefit from lower pressures, while those with LV-predominant heart failure benefit from higher pressures.1

If the patient is unable to tolerate a face mask, does not yet require (or refuses) endotracheal intubation, but remains hypoxic, high flow nasal cannula (HFNC) may be considered. HFNC has not been exclusively studied in CS, but it has been shown to improve oxygenation enough to prevent intubation in severe cardiogenic pulmonary edema.18 For the severely anxious patient, HFNC is more comfortable, better tolerated, causes less skin breakdown, and will still have the desired effect of decreasing RR and increasing oxygenation.19 HFNC can be administered to a maximum of 60 L/min, and may provide up to around 7 cmH2O of PEEP.1 Notably, HFNC will not have any marked effect on hypercapnia, so if pCO2 is elevated, BiPAP or endotracheal intubation is preferable.19 Additionally, HFNC is not hemodynamically neutral and should not be used to avoid the hemodynamic effects of positive pressure ventilation– in some cases, it has a hemodynamic impact comparable to NIPPV.19
// C – Circulation
Goal MAP may differ slightly between patients depending on their baseline blood pressure, but a reasonable goal is 65-70 mm Hg.20 A target MAP >70 mm Hg may be harmful unless the patient is chronically hypertensive. 20 In the case of chronic hypertension, the target MAP should be established based on signs of adequate perfusion, including mentation, urine output, and temperature to guide the titration of vasopressors/inotropes.21 Once a goal MAP range is established, care should be taken to remain within this range to avoid unnecessarily increasing myocardial oxygen demand and the frequency of dysrhythmias by overshooting the mark.9 If the patient is especially sensitive to changes in MAP and it is hard to remain in range, overshooting is better than undershooting.22
To maintain adequate perfusion, the general strategy in CS is to start with an inopressor, which increases systemic vascular resistance (SVR) and inotropy (Table 3). If the patient has any signs of poor cardiac output or hypoperfusion, an inodilator – which increases inotropy but decreases SVR – should be added.13 Inodilators are never first-line in CS because they can produce profound vasodilation when given alone.13 While there continues to be debate, norepinephrine (NE) is often used as the first-line inopressor for CS.20 The effects of NE are primarily α1, followed by some β1 and even less β2, therefore its effects are primarily to increase SVR and cardiac contractility.21 NE has been shown in animal models to increase CI without increasing HR or serum lactate.24 In a multicenter, randomized study of patients with CS, NE outperformed dopamine, with a decreased death rate and fewer adverse effects in the NE group.23
Vasopressin is an agonist at V1 receptors in vascular smooth muscle, and it may be added as an adjunct to NE to help increase SVR.21 It should be used judiciously in patients with primary left-heart dysfunction, as it increases left ventricular afterload without augmenting contractility. This drug is helpful in patients with severe pulmonary hypertension since it does not increase pulmonary vascular resistance.9 Unfortunately, evidence for its use in cardiogenic shock is lacking with no randomized control trials in that particular context being available at this time. Epinephrine has strong α1, β1 and β2 agonism, but is not routinely recommended in CS.25,26 Epinephrine causes comparable increases in MAP and cardiac index in CS when compared to NE, but it may also increase the likelihood of refractory shock.25 Epinephrine may also increase the risk of mortality in patients with CS. 26 Compared to epinephrine, a norepinephrine/dobutamine combination generates higher CI in patients with CS without a concomitant increase in HR and without raising serum lactate levels.27 Dopamine has fallen out of favor in the context of CS and is not recommended due to it increasing oxygen consumption and the risk for dysrhythmias when given alone.20
Notably, CS is unique compared with other types of hypoperfusion states in that its manifestations are in part due to failure of the pump itself, so augmenting function of the heart itself plays a role in the treatment of CS. Pirracchio et al. found that adding an inodilator (e.g. dobutamine or PD-3 inhibitor) to a vasopressor in CS was associated with decreased short-term mortality compared to use of vasopressors alone.28 For patients with very poor LVEF, dobutamine can be considered as an adjunct to norepinephrine.20,21 Dobutamine’s effects are primarily agonism at β1, leading to increased contractility and increased stroke volume without a concurrent increase in SVR.2 It should be started at a low dose, such as 2.0 mcg/kg/min, and carefully titrated to effect.20 Excessively high doses will increase the size of an acute infarct (if present) due to the upsurge in myocardial oxygen demand and wall stress.2
Another inotrope to consider instead of dobutamine is milrinone, a PD-3 inhibitor, which increases inotropy and decreases SVR.29 Opportunities to use milrinone have been somewhat limited in light of the OPTIME-CHF trial (2003), which showed worse outcomes when it was used in CS secondary to an ischemic event.30 However, the recent DOREMI (2021) randomized clinical trial comparing the use of milrinone to dobutamine in patients with CS showed no difference between in-hospital death rates, need for mechanical circulatory support, or need for renal replacement therapy.29 Some suggest a combination of milrinone with vasopressin to counteract the decreased SVR, especially in patients who are insensitive to catecholamines due to chronic beta-blocker (BB) therapy.9,20
Table 3. Vasopressor and inodilator options in CS.

Additional Testing
In addition to a physical examination and history, the following studies may aid in the diagnosis of CS and elucidation of its underlying cause:
- CBC, chemistry, liver function tests, ionized calcium, magnesium, phosphorous, troponin, brain-type natriuretic peptide (BNP) or N-terminal pro-brain-type natriuretic peptide (NT-proBNP), lactate ABG
- Urinalysis
- ECG
- Bedside echo
- Chest X-ray
Management
It may be helpful to categorize etiologies of CS by their underlying cause (e.g. Structure, Squeeze, and Stoppage) (Figure 3).
Figure 3. Causes of CS organized by primary problem. This is not all-inclusive but covers many of the common etiologies.

Structure
Table 4 summarizes the structural causes of cardiogenic shock and considerations for their treatment. Each cause is discussed in more detail below.
Table 4. Structural causes of CS.

Acute Mitral Regurgitation
Diagnostic clues: Holosystolic murmur loudest at the apex that worsens with straining, acute MI (AMI) within the last seven days.
Goal(s): (Carefully) increase afterload and early surgical consultation.
Acute mitral regurgitation (AMR) often occurs as a complication of an AMI two to seven days after the primary injury.31 The most common cause is the rupture of the posteromedial papillary muscle due to its singular blood supply from either the right coronary or left circumflex artery.31 Other causes for AMR include leaflet perforation in endocarditis, leaflet inflammation in rheumatic carditis, Takotsubo cardiomyopathy, and peripartum cardiomyopathy.31Norepinephrine is the first-line vasopressor in mitral regurgitation. Once stabilized with a vasopressor, an inotrope should be added. These patients are sensitive to increased afterload and may benefit from early intra-aortic balloon pump (IABP) titrated to the lowest possible perfusing pressure.21 Early involvement of a cardiothoracic surgeon is critical, as mortality is close to 80% without repair of the mitral valve (if surgical repair is indicated, as in AMI).31
Ventricular Septal Rupture
Diagnostic clues: Sudden onset of CS, new holosystolic murmur, RV dilatation or Doppler flow between ventricles on ultrasound.32
Goal(s): Afterload reduction and early surgical consultation.
Ventricular septal rupture (VSR) is a complication of 0.3% of acute STEMI with a 40-80% mortality rate.33 The goal of ED treatment in VSR is the reduction of afterload to decrease the left-to-right shunt. The combination of an inotrope with a vasodilator (nitroprusside) appears to be the best combination to reduce shunting. Vasopressors or inotropes alone increase left-to-right shunting, and possibly afterload. Mechanical circulatory support devices such as ImpellaTM [optimally effective when combined with extracorporeal membrane oxygenation (ECMO)] are often used as a bridge to surgical repair or transplant.33
Free Wall Rupture
Diagnostic clues: AMI in the last seven days, pericardial effusion on ultrasound.
Goal(s): Optimize preload and early surgical consultation.
Rupture of the LV after an AMI is often immediately fatal, but CS is the presenting syndrome in those who survive.34 This complication occurs within the first week after an AMI, when the myocardium is the weakest.35 Pericardial effusion is almost invariably present on ultrasound; >5 mm effusion on ultrasound has been demonstrated to have 100% sensitivity for subacute free wall rupture in patients with subacute MI.34 Definitive treatment is surgical relief of the tamponade and repair of the ventricular wall defect.35 IABP is often used to help stabilize hemodynamics prior to surgery but has mixed results. VA-ECMO has no consistent data showing improved outcomes in these patients.36 It is probably best to leave pericardiocentesis for the operating room, since opening the pericardial sac may lead to hemorrhagic shock.34 Pericardiocentesis may be attempted if cardiothoracic surgery is not immediately available (rural ED) and medical therapy is not working, but in most cases this is not recommended.74
RV Failure
Diagnostic clues: Tricuspid regurgitation, dependent edema, right atrial (RA)/RV dilatation on ultrasound, right axis deviation/right bundle branch block/RV hypertrophy/right heart strain on ECG, enlarged RA/RV on chest X-ray. 37,38
Goal(s): Optimize preload, aggressive treatment of hypoxemia, and early reperfusion [if AMI or pulmonary embolism (PE)].
Acute RV failure is often secondary to AMI or massive pulmonary embolism. RV dilatation causes bowing of the interventricular septum into the LV, decreasing LV cardiac output. In addition, tricuspid regurgitation from lack of forward flow leads to venous congestion. In addition to emergent revascularization when indicated, mainstays of treatment include optimizing preload and cautious use of inotropes.39 Ventilator settings should aim to aggressively treat hypoxemia; this will decrease hypoxic pulmonary vasoconstriction and thus RV afterload.40 PEEP settings should start at 3-5 mmHg.1 When possible, inspiratory time should be kept short, minimizing mean airway pressures and RV afterload.41 Patients with RV infarct may be profoundly bradycardic and require external pacing. Norepinephrine with dobutamine is the preferred vasopressor/inotrope combination.42 Unfractionated heparin is the preferred anticoagulation choice in CS with RV infarct or PE.42
Aortic Dissection
Diagnostic clues: Abrupt, severe, tearing, migrating chest/back/abdominal pain, pulse deficit, neurologic deficit, pericardial effusion.43
Goal(s): Control HR/BP, control pain, and early surgical consultation.
Stanford Type A aortic dissection (which, by definition, involves the ascending aorta) is most likely to cause cardiac complications, including CS. A Type A dissection is associated with aortic regurgitation, cardiac tamponade, and acute myocardial ischemia from compression of the coronary arterial ostium.44,45 Typical treatment for acute aortic dissection is aggressive control of pain, BP, and HR. Type A dissections require emergent surgical repair. Goal SBP is between 100-120 mm Hg and goal HR is 60 bpm to minimize shear stress on the aortic wall and prevent worsening of the dissection.45
Acute Aortic Regurgitation
Diagnostic clues: High-pitched blowing diastolic murmur loudest at left third/fourth intercostal space.
Goal(s): Reduce afterload and early surgical consultation.
Most often acute aortic regurgitation (AAR) is secondary to endocarditis, blunt trauma, or aortic dissection. The ultimate treatment for AAR is emergent surgical correction, but a vasodilator like nitroglycerin or nitroprusside may be combined with an inotrope like dobutamine to help with afterload reduction and squeeze. IABP is contraindicated in AAR, because diastolic inflation increases regurgitation of blood into the LV.46
Squeeze
Table 5 summarizes the causes of cardiogenic shock secondary to impaired contractility and considerations for their treatment. Each cause is discussed in more detail below.
Table 5. Causes of impaired contractility (“Squeeze”) leading to CS.

Sepsis-Induced Cardiomyopathy
Diagnostic clues: Fever, known or suspected source of infection.
Goal(s): Volume resuscitation, early antibiotics, infectious source control.
The pathophysiology of sepsis-induced CS is not completely understood, but Habimana et al. (2020) present a strong argument for the triad of (1) direct myocardial suppression, (2) intrinsic myocardial circulatory dysfunction, and (3) mitochondrial dysfunction.47 This type of cardiomyopathy is often defined by its acute nature, absence of acute coronary syndrome (ACS), and reversibility after the primary insult is resolved.48
Sepsis-induced CS presents with some or (often) all of the following: LV dysfunction, RV dysfunction, and dilation of both ventricles.47 In addition, patients with sepsis-induced CS will often fail to respond to IV fluid challenge or initial vasopressors.48 Treatment in the ED includes rapid initiation of broad-spectrum antibiotic therapy, identification and control of the infectious source, and IV fluids. Importantly, IV fluids must be given judiciously, as these patients are at risk of pulmonary edema from both LV failure and abnormally high permeability of pulmonary capillary beds.49 Norepinephrine remains the first-line vasopressor if the patient is unresponsive to fluids.49
Myocarditis
Diagnostic clues: Distinct onset of symptoms, viral prodrome, elevated troponin, ischemic ECG changes.
Goal(s): Volume optimization and treatment of infection
Myocarditis is a difficult ED diagnosis, but an important cause of CS to consider. It is primarily a disease of young adults and pediatric patients, although it can occur at any age.50 Clinical presentation is variable, with the most common symptoms being dyspnea and chest pain.51 Key historical features include a distinct symptom onset within two to four weeks prior to presentation and a preceding viral prodrome since viral infections are the most common cause of myocarditis.50 These patients may have elevated troponin I levels and ischemic ECG changes, but non-steroidal anti-inflammatory drugs (NSAIDs) are not recommended because they can worsen myocardial damage.51 In addition, these patients may be resistant to vasopressor and inodilator therapy. They may need mechanical support such as IABP or LV assist devices.51 Failure of these devices to stabilize CS may necessitate transfer to a transplant center.51
Medication Toxicity/Poisoning
Diagnostic clues: Bradycardia resistant to chronotropes, known or suspected toxic ingestion.
Goal(s): Decontamination, toxin-specific treatment.
Many different drug classes have cardiotoxic effects in high doses, including Class I antiarrhythmics, BB, calcium channel blockers (CCB), tricyclic antidepressants (TCA), selective serotonin reuptake inhibitors (SSRIs), dopamine and norepinephrine reuptake inhibitors (like bupropion), anti-epileptics (carbamazepine, phenytoin), phenothiazines, opioids, antimalarial drugs (chloroquine), and cocaine.52 A few of these deserve special mention.
BB and CCB overdose should be treated with high-dose insulin euglycemic therapy, a trial of high-dose glucagon, calcium, IV crystalloids, and potentially cardiac pacing, as needed.53 Decontamination via gastric lavage or whole-bowel irrigation is something to consider if ingestion was within one hour of arrival, but these procedures significantly increase the risk of vomiting and aspiration in non-intubated patients.54 CS is especially insensitive to vasopressors and inotropes in CCB/BB overdose according to multiple animal studies and human case reports.55 Vasopressors appear to work better in the context of high-dose insulin therapy.55 The patient may require some sort of mechanical support such as IABP or ECMO. Lipid emulsion therapy may also be considered but should be avoided if planning to attempt hemodialysis to clear certain dialyzable BB (atenolol).56
TCA toxicity depresses myocardial function by slowing inward sodium current during depolarization and causing peripheral vasodilation by direct α-antagonism. First-line treatment is sodium bicarbonate which both overwhelms the sodium blockade and alkalinizes the blood, reducing TCA binding to sodium channels. Epinephrine may be more effective than norepinephrine in this context.57
Carbon monoxide causes tissue hypoxia and is directly cardiotoxic. First-line therapy for carbon monoxide poisoning is the administration of 100% FiO2 via non-rebreather, NIPPV, or endotracheal tube (ETT), and hyperbaric oxygen chamber if clinically indicated.58 The typical vasopressor/inodilator combinations described above are still recommended, but they may exacerbate the hyperadrenergic state induced by carbon monoxide.58
Myocardial Contusion
Diagnostic clues: Blunt chest trauma with elevated cardiac troponin levels, ischemic ECG changes, hypokinetic wall segment on ultrasound.
Goal(s): Optimize preload, control/treat active bleeding, and evaluate for tamponade.
Myocardial contusions are the result of a direct high-energy impact to the chest and are a rare cause of CS. The RV is the most susceptible due to its anterior location within the chest cavity.59 Therefore, these lesions often create preload dependence, which has consequences in acutely hypovolemic and mechanically-ventilated patients. Treatment should be focused on volume optimization with either IV crystalloids or blood products, and using low PEEP to support RV function.59
Unstable Arrhythmia
Diagnostic clues: Provided by the ECG or cardiac monitor.
Goal(s): Control the inciting rhythm.
For any unstable arrhythmia causing CS, care should be focused on terminating the arrhythmia and then searching for the underlying cause. For unstable tachyarrhythmias, electrical cardioversion is first-line.60 Special focus should be on correcting electrolyte abnormalities (especially magnesium and potassium) and treating thyrotoxicosis (if present). CCB are contraindicated in CS, so if pharmacological cardioversion is desired for atrial fibrillation with rapid ventricular rate (RVR), amiodarone (150 mg/10 min, then 1 mg/min for six hours, then 0.5 mg/min for 18 hours) or digoxin (0.25 mg IV bolus and then 0.75-0.15 mg over 24 hours) may be used unless a pre-excitation syndrome is suspected (in which case procainamide is indicated).60 Digoxin use as an antiarrhythmic should be done in consultation with cardiology. In the case of sustained VT or repeated episodes of ventricular fibrillation (VF), amiodarone may be used, and BB should be avoided. These patients in particular may require mechanical support with IABP, Impella,TM or ECMO.60
For unstable bradyarrhythmias, transcutaneous or transvenous pacing should predominate.60 Recommended pharmaceutical interventions include atropine, isoproterenol, dobutamine, and epinephrine. Consider the possibility of BB or CCB toxicity, electrolyte derangement, or myxedema as possible causes, especially if the rhythm is refractory to conventional treatment.21
AMI
Diagnostic clues: Elevated cardiac troponin levels > 99th percentile upper limit of normal, new ischemic changes on ECG, new pathologic Q waves, ischemic symptoms (chest pain, dyspnea, fatigue), new wall motion abnormality on ultrasound.61
Goal(s): Early recognition, early reperfusion, and/or early transfer to PCI center.
AMI is the primary cause for 70% of all cases of CS.13 CS is more likely to result from ST-elevation myocardial infarction (STEMI) than non-STEMI (NSTEMI).2 For patients with confirmed AMI causing CS, definitive care is reperfusion therapy.21 This is accomplished by some or all of the following: fibrinolysis, cardiac catheterization with percutaneous coronary intervention (PCI), and coronary artery bypass graft (CABG).21 Of these three, fibrinolysis is inferior in CS, possibly because fibrinolytics require adequate perfusion pressure to be circulated appropriately.21 The SHOCK trial compared fibrinolysis plus IABP to early PCI or CABG plus IABP in patients presenting with CS from AMI and found that the latter was superior in terms of short- and long-term mortality. Of those in the PCI group, lower mortality was seen in those treated with stents than those treated with balloon angioplasty. Further, those who received PCI by radial artery access had fewer complications and decreased mortality than those who received PCI through the femoral artery.4 Emergency physicians should ensure the structural integrity of the right radial artery for the cardiologist if possible. If PCI cannot occur within 120 minutes of the AMI, fibrinolysis should be given.
Antiplatelet agents and heparin are the standard treatments for AMI, but there are nuances in the case of CS. CS, like all hypoperfusion states, decreases enteral blood flow, and as a result, absorption of drugs given orally (or rectally) is decreased.62 Platelet inhibition is therefore also diminished in CS unless the drugs are given parenterally. Current American Heart Association (AHA) guidelines recommend the use of intravenous (IV) abciximab (a glycoprotein IIb/IIIa inhibitor) or cangrelor (a P2Y12 inhibitor) if intestinal absorption is a concern;21 however, the IABP-SHOCK trial found no significant difference in mortality between clopidogrel, prasugrel, and ticagrelor among patients with CS secondary to AMI.5 Based on what little research exists, there does not appear to be a decrease of the antiplatelet effect of aspirin in shock states, so it may be given in standard dosing/route.62 Current recommendations endorse the use of unfractionated heparin (UFH) over low-molecular-weight heparin (LMWH) to avoid unnecessary stress on the kidneys in AMI.21
Stoppage
Table 6 summarizes the obstructive causes of cardiogenic shock secondary and considerations for their treatment. Each cause is discussed in more detail below.
Table 6. Obstructive (“stoppage”) causes of CS.
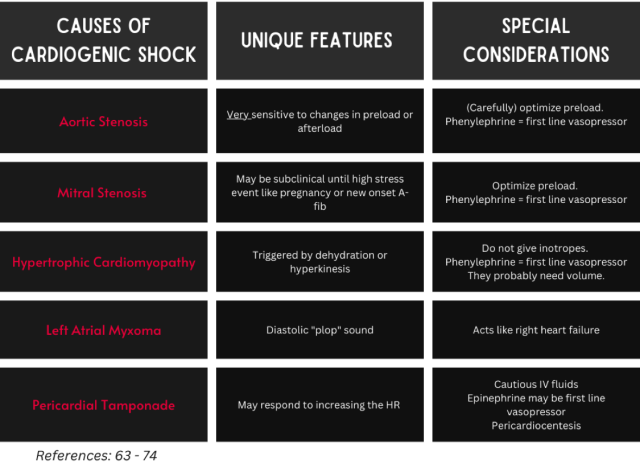
Critical Aortic Stenosis
Diagnostic clues: High-pitched mid-systolic ejection murmur loudest at the right upper sternal border, pulsus parvus et tardus (weakened/delayed carotid pulse), recent (especially exercise-induced) syncope.63
Goal(s): Avoid unnecessary increases in afterload, as this hinders diastolic coronary perfusion.64,65 Optimize preload (carefully).
Consider using phenylephrine as the initial vasopressor to avoid the reduced diastolic filling time caused by tachycardia. The alpha-mediated increase in afterload is fixed at the level of the aortic valve, and the resultant increase in SVR can induce that reflex bradycardia that can improve supply/demand mismatch by augmenting cardiac filling. Norepinephrine should be used if a small increase in inotropy is needed. Vasodilators should be avoided as they will worsen preload. If inotropes are needed, use the lowest dose possible.66
When this condition becomes symptomatic, the definitive treatment is valve replacement.64 Patients with CS often require balloon aortic valvuloplasty and/or mechanical circulatory support (i.e. IABP, ECMO) as temporizing measures before surgical replacement because they are too unstable for the operating room.67
Mitral Stenosis
Diagnostic clues: Diastolic opening snap and low-pitched decrescendo-crescendo diastolic murmur.68
Goal(s): Optimize preload and slow tachycardias.
This is a preload-dependent state, so cautious IV crystalloid administration is warranted. If vasopressors are required, phenylephrine or vasopressin are preferred. Amiodarone or esmolol may be used to slow HR and allow for adequate diastolic filling time.21
Definitive treatment is balloon mitral valvuloplasty, but outcomes in the context of CS are poor.69
Hypertrophic Cardiomyopathy
Diagnostic clues: Episodes of syncope, crescendo-decrescendo systolic ejection murmur loudest at the left sternal border that gets better with straining.70
Goal(s): Increase preload, slow tachycardias, and increase afterload.71
This is the most common cause of LVOT obstruction (LVOTO). Another notable cause includes Takotsubo cardiomyopathy. LVOTO is exacerbated by volume depletion and increased inotropy, such as in sepsis, dehydration, or anemia. Therefore, treatment involves “filling the tank” – volume repletion with IV crystalloids or blood if indicated – and increasing SVR. The first-line vasopressor is phenylephrine, followed by vasopressin, and lastly norepinephrine. Inotropes are contraindicated, as they exacerbate dynamic LVOTO.72 Vasodilators should likewise be avoided, since they worsen preload.21
Left Atrial (LA) Myxoma
Diagnostic clues: Diastolic “tumor plop” sound, LA enlargement on ECG, cardiomegaly on chest X-ray, signs of right heart failure, may present with stroke symptoms.
Goal(s): Optimize preload and early surgical consultation.
The vast majority of cardiac myxomas are in the LA, and can cause CS when they obstruct the mitral valve and lead to right heart failure. Definitive treatment is surgical removal.73
Pericardial Tamponade
Diagnostic clues: Pulsus paradoxus, low voltage or electrical alternans on ECG, pericardial effusion with diastolic RV collapse, systolic RA collapse, plethoric IVC on ultrasound.74,75
Goal(s): Careful volume repletion, (possibly) HR elevation, and pericardiocentesis.
Some patients may require volume repletion in the context of severe tamponade to help prevent right heart collapse, but this should be done carefully as these patients can become volume-overloaded quickly. Epinephrine may be favored over norepinephrine for the first-line vasopressor if increased HR is needed to increase cardiac output since stroke volume is limited. Pericardiocentesis is the definitive treatment and should be attempted (with ultrasound guidance) in emergent situations in the ED.74
Case Conclusion:
The patient’s chest X-ray showed bilateral pleural effusions, extensive pulmonary edema, and pulmonary hilar hypervascularity. Bedside echo showed akinesis of the apex and anterior wall of the heart, mitral regurgitation, and a sclerotic aortic valve. Laboratory workup revealed mild leukocytosis, elevated troponin, elevated BNP, mild transaminitis, hyperglycemia, and elevated lactate. The patient was switched to BiPAP, started on an amiodarone drip, given empiric antibiotics, and diuresed with furosemide. His condition improved and he was admitted to the ICU. In the ICU, he was started on dobutamine and furosemide drips. He was discharged home in stable condition after one week in the hospital.
Pearls and Pitfalls
- Recent MI is the most common cause of CS.
- Norepinephrine is often the vasopressor of choice in CS unless a significant contraindication exists.
- The RV benefits from lower PEEP. The LV benefits from higher PEEP.
- NIPPV/HFNC are the first-line interventions for respiratory distress in CS, but if the patient needs to be intubated, the use of hemodynamically-stable induction agents such as etomidate and ketamine is advised.
- It is important to have an idea of the primary cause of CS since treatment varies by etiology.
- Avoid CCB in all forms of CS.
References:
(1) Alviar, C. L.; Miller, P. E.; McAreavey, D.; Katz, J. N.; Lee, B.; Moriyama, B.; Soble, J.; van Diepen, S.; Solomon, M. A.; Morrow, D. A. Positive Pressure Ventilation in the Cardiac Intensive Care Unit. Journal of the American College of Cardiology 2018, 72 (13), 1532-1553. DOI: https://doi.org/10.1016/j.jacc.2018.06.074.
(2) Westaby, S.; Kharbanda, R.; Banning, A. P. Cardiogenic shock in ACS. Part 1: prediction, presentation and medical therapy. Nat Rev Cardiol 2011, 9 (3), 158-171. DOI: 10.1038/nrcardio.2011.194 From NLM.
(3) Reyentovich, A.; Barghash, M. H.; Hochman, J. S. Management of refractory cardiogenic shock. Nat Rev Cardiol 2016, 13 (8), 481-492. DOI: 10.1038/nrcardio.2016.96 From NLM.
(4) Hochman, J. S.; Buller, C. E.; Sleeper, L. A.; Boland, J.; Dzavik, V.; Sanborn, T. A.; Godfrey, E.; White, H. D.; Lim, J.; LeJemtel, T. Cardiogenic shock complicating acute myocardial infarction–etiologies, management and outcome: a report from the SHOCK Trial Registry. SHould we emergently revascularize Occluded Coronaries for cardiogenic shocK? J Am Coll Cardiol 2000, 36 (3 Suppl A), 1063-1070. DOI: 10.1016/s0735-1097(00)00879-2 From NLM.
(5) Thiele, H.; Zeymer, U.; Neumann, F. J.; Ferenc, M.; Olbrich, H. G.; Hausleiter, J.; Richardt, G.; Hennersdorf, M.; Empen, K.; Fuernau, G.; et al. Intraaortic balloon support for myocardial infarction with cardiogenic shock. N Engl J Med 2012, 367 (14), 1287-1296. DOI: 10.1056/NEJMoa1208410 From NLM.
(6) Dinh, V. A.; Ko, H. S.; Rao, R.; Bansal, R. C.; Smith, D. D.; Kim, T. E.; Nguyen, H. B. Measuring cardiac index with a focused cardiac ultrasound examination in the ED. Am J Emerg Med 2012, 30 (9), 1845-1851. DOI: 10.1016/j.ajem.2012.03.025 From NLM.
(7) Shaikh, F.; Kenny, J.-E.; Awan, O.; Markovic, D.; Friedman, O.; He, T.; Singh, S.; Yan, P.; Qadir, N.; Barjaktarevic, I. Measuring the accuracy of cardiac output using POCUS: the introduction of artificial intelligence into routine care. The Ultrasound Journal 2022, 14 (1), 47. DOI: 10.1186/s13089-022-00301-6.
(8) Polcz, M.; Huston, J.; Breed, M.; Case, M.; Leisy, P.; Schmeckpeper, J.; Vaughn, L.; Sobey, J. H.; Brophy, C.; Lindenfeld, J.; et al. Comparison of clinical symptoms and bioimpedance to pulmonary capillary wedge pressure in heart failure. American Heart Journal Plus: Cardiology Research and Practice 2022, 15, 100133. DOI: https://doi.org/10.1016/j.ahjo.2022.100133.
(9) Chioncel, O.; Parissis, J.; Mebazaa, A.; Thiele, H.; Desch, S.; Bauersachs, J.; Harjola, V. P.; Antohi, E. L.; Arrigo, M.; Ben Gal, T.; et al. Epidemiology, pathophysiology and contemporary management of cardiogenic shock – a position statement from the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 2020, 22 (8), 1315-1341. DOI: 10.1002/ejhf.1922 From NLM.
(10) Forrester, J.; Diamond, G.; Chatterjee, K.; Swan, H. Medical Therapy of Acute Myocardial Infarction by Application of Hemodynamic Subsets. The New England journal of medicine 1977, 295, 1356-1362. DOI: 10.1056/NEJM197612092952406.
(11) Daly, M.; Lentz, S. Diagnosing Cardiogenic Shock in the ED. In emDocs, Montrief, T., Koyfman, A., Long, B., Eds.; 2020; Vol. 2022.
(12) Reim, P.; Moore, L.; Minalyan, A.; Dinh, V. RUSH Exam Ultrasound Protocol In POCUS 101, Dinh, V., Ed.; Vol. 2022.
(13) Daly, M.; Long, B.; Koyfman, A.; Lentz, S. Identifying cardiogenic shock in the emergency department. Am J Emerg Med 2020, 38 (11), 2425-2433. DOI: 10.1016/j.ajem.2020.09.045 From NLM.
(14) McDonagh, T. A.; Metra, M.; Adamo, M.; Gardner, R. S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: Developed by the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) With the special contribution of the Heart Failure Association (HFA) of the ESC. European Heart Journal 2021, 42 (36), 3599-3726. DOI: 10.1093/eurheartj/ehab368 (acccessed 12/21/2022).
(15) Masip, J.; Peacock, W. F.; Price, S.; Cullen, L.; Martin-Sanchez, F. J.; Seferovic, P.; Maisel, A. S.; Miro, O.; Filippatos, G.; Vrints, C.; et al. Indications and practical approach to non-invasive ventilation in acute heart failure. Eur Heart J 2018, 39 (1), 17-25. DOI: 10.1093/eurheartj/ehx580 From NLM.
(16) Hongisto, M.; Lassus, J.; Tarvasmaki, T.; Sionis, A.; Tolppanen, H.; Lindholm, M. G.; Banaszewski, M.; Parissis, J.; Spinar, J.; Silva-Cardoso, J.; et al. Use of noninvasive and invasive mechanical ventilation in cardiogenic shock: A prospective multicenter study. Int J Cardiol 2017, 230, 191-197. DOI: 10.1016/j.ijcard.2016.12.175 From NLM.
(17) Vahdatpour, C.; Collins, D.; Goldberg, S. Cardiogenic Shock. Journal of the American Heart Association 2019, 8 (8), e011991. DOI: doi:10.1161/JAHA.119.011991.
(18) Kang, M. G.; Kim, K.; Ju, S.; Park, H. W.; Lee, S. J.; Koh, J. S.; Hwang, S. J.; Hwang, J. Y.; Bae, J. S.; Ahn, J. H.; et al. Clinical efficacy of high-flow oxygen therapy through nasal cannula in patients with acute heart failure. J Thorac Dis 2019, 11 (2), 410-417. DOI: 10.21037/jtd.2019.01.51 From NLM.
(19) Hernández, G.; Roca, O.; Colinas, L. High-flow nasal cannula support therapy: new insights and improving performance. Critical Care 2017, 21 (1), 62. DOI: 10.1186/s13054-017-1640-2.
(20) Levy, B.; Bastien, O.; Bendjelid, K.; Cariou, A.; Chouihed, T.; Combes, A.; Mebazaa, A.; Megarbane, B.; Plaisance, P.; Ouattara, A.; et al. Experts’ recommendations for the management of adult patients with cardiogenic shock. Annals of Intensive Care 2015, 5 (1). DOI: 10.1186/s13613-015-0052-1.
(21) van Diepen, S.; Katz, J. N.; Albert, N. M.; Henry, T. D.; Jacobs, A. K.; Kapur, N. K.; Kilic, A.; Menon, V.; Ohman, E. M.; Sweitzer, N. K.; et al. Contemporary Management of Cardiogenic Shock: A Scientific Statement From the American Heart Association. Circulation 2017, 136 (16), e232-e268. DOI: 10.1161/cir.0000000000000525 From NLM.
(22) Burstein, B.; Tabi, M.; Barsness, G. W.; Bell, M. R.; Kashani, K.; Jentzer, J. C. Association between mean arterial pressure during the first 24 hours and hospital mortality in patients with cardiogenic shock. Crit Care 2020, 24 (1), 513. DOI: 10.1186/s13054-020-03217-6 From NLM.
(23) De Backer, D.; Biston, P.; Devriendt, J.; Madl, C.; Chochrad, D.; Aldecoa, C.; Brasseur, A.; Defrance, P.; Gottignies, P.; Vincent, J.-L. Comparison of Dopamine and Norepinephrine in the Treatment of Shock. New England Journal of Medicine 2010, 362 (9), 779-789. DOI: 10.1056/NEJMoa0907118.
(24) Beurton, A.; Ducrocq, N.; Auchet, T.; Joineau-Groubatch, F.; Falanga, A.; Kimmoun, A.; Girerd, N.; Fay, R.; Vanhuyse, F.; Tran, N.; et al. Beneficial Effects of Norepinephrine Alone on Cardiovascular Function and Tissue Oxygenation in a Pig Model of Cardiogenic Shock. Shock 2016, 46 (2), 214-218. DOI: 10.1097/shk.0000000000000579 From NLM.
(25) Levy, B.; Clere-Jehl, R.; Legras, A.; Morichau-Beauchant, T.; Leone, M.; Frederique, G.; Quenot, J. P.; Kimmoun, A.; Cariou, A.; Lassus, J.; et al. Epinephrine Versus Norepinephrine for Cardiogenic Shock After Acute Myocardial Infarction. J Am Coll Cardiol 2018, 72 (2), 173-182. DOI: 10.1016/j.jacc.2018.04.051 From NLM.
(26) Léopold, V.; Gayat, E.; Pirracchio, R.; Spinar, J.; Parenica, J.; Tarvasmäki, T.; Lassus, J.; Harjola, V. P.; Champion, S.; Zannad, F.; et al. Epinephrine and short-term survival in cardiogenic shock: an individual data meta-analysis of 2583 patients. Intensive Care Med 2018, 44 (6), 847-856. DOI: 10.1007/s00134-018-5222-9 From NLM.
(27) Levy, B.; Perez, P.; Perny, J.; Thivilier, C.; Gerard, A. Comparison of norepinephrine-dobutamine to epinephrine for hemodynamics, lactate metabolism, and organ function variables in cardiogenic shock. A prospective, randomized pilot study. Crit Care Med 2011, 39 (3), 450-455. DOI: 10.1097/CCM.0b013e3181ffe0eb From NLM.
(28) Pirracchio, R.; Parenica, J.; Resche Rigon, M.; Chevret, S.; Spinar, J.; Jarkovsky, J.; Zannad, F.; Alla, F.; Mebazaa, A.; for the, G. n. The Effectiveness of Inodilators in Reducing Short Term Mortality among Patient with Severe Cardiogenic Shock: A Propensity-Based Analysis. PLOS ONE 2013, 8 (8), e71659. DOI: 10.1371/journal.pone.0071659.
(29) Mathew, R.; Di Santo, P.; Jung, R. G.; Marbach, J. A.; Hutson, J.; Simard, T.; Ramirez, F. D.; Harnett, D. T.; Merdad, A.; Almufleh, A.; et al. Milrinone as Compared with Dobutamine in the Treatment of Cardiogenic Shock. New England Journal of Medicine 2021, 385 (6), 516-525. DOI: 10.1056/NEJMoa2026845.
(30) Felker, G. M.; Benza, R. L.; Chandler, A. B.; Leimberger, J. D.; Cuffe, M. S.; Califf, R. M.; Gheorghiade, M.; O’Connor, C. M. Heart failure etiology and response to milrinone in decompensated heart failure: results from the OPTIME-CHF study. J Am Coll Cardiol 2003, 41 (6), 997-1003. DOI: 10.1016/s0735-1097(02)02968-6 From NLM.
(31) Briosa, E. G. A.; Hinton, J.; Sirohi, R. Cardiogenic shock due to acute severe ischemic mitral regurgitation. Am J Emerg Med 2021, 43, 292.e291-292.e293. DOI: 10.1016/j.ajem.2020.10.028 From NLM.
(32) Farkas, J. Post-MI Complications. In The Internet Book of Critical Care, Farkas, J., Ed.; 2021; Vol. 2022.
(33) Pahuja, M.; Schrage, B.; Westermann, D.; Basir, M. B.; Garan, A. R.; Burkhoff, D. Hemodynamic Effects of Mechanical Circulatory Support Devices in Ventricular Septal Defect. Circulation: Heart Failure 2019, 12 (7), e005981. DOI: doi:10.1161/CIRCHEARTFAILURE.119.005981.
(34) Slater, J.; Brown, R. J.; Antonelli, T. A.; Menon, V.; Boland, J.; Col, J.; Dzavik, V.; Greenberg, M.; Menegus, M.; Connery, C.; et al. Cardiogenic shock due to cardiac free-wall rupture or tamponade after acute myocardial infarction: a report from the SHOCK Trial Registry. Journal of the American College of Cardiology 2000, 36 (3, Supplement 1), 1117-1122. DOI: https://doi.org/10.1016/S0735-1097(00)00845-7.
(35) Mishra, P. K.; Pathi, V.; Murday, A. Post myocardial infarction left ventricular free wall rupture. Interactive CardioVascular and Thoracic Surgery 2007, 6 (1), 39-42. DOI: 10.1510/icvts.2006.138511 (accessed 12/25/2022).
(36) Matteucci, M.; Formica, F.; Kowalewski, M.; Massimi, G.; Ronco, D.; Beghi, C.; Lorusso, R. Meta-analysis of surgical treatment for postinfarction left ventricular free-wall rupture. J Card Surg 2021, 36 (9), 3326-3333. DOI: 10.1111/jocs.15701 From NLM.
(37) Vallabhajosyula, S.; Kumar, M.; Pandompatam, G.; Sakhuja, A.; Kashyap, R.; Kashani, K.; Gajic, O.; Geske, J. B.; Jentzer, J. C. Prognostic impact of isolated right ventricular dysfunction in sepsis and septic shock: an 8-year historical cohort study. Ann Intensive Care 2017, 7 (1), 94. DOI: 10.1186/s13613-017-0319-9 From NLM.
(38) Ray, K.; Parmar, M.; McCloskey, C. Pulmonary Hypertension in the ED In NUEM, McCloskey, C., Ed.; Vol. 2022.
(39) Salam, M. F.; Gorgis, S.; Basir, M. B. Current and Emerging Strategies for RV Shock Management in the Setting of RV Infarct. In Latest in Cardiology, American College of Cardiology 2021.
(40) Held, N.; Little, N.; Krantz, M. J.; Stauffer, B. L. Refractory Cardiogenic Shock from Right Ventricular Infarction Successfully Managed with Inhaled Epoprostenol. Am J Case Rep 2017, 18, 271-275. DOI: 10.12659/ajcr.901975 From NLM.
(41) Uddin, S.; Anandanadesan, R.; Trimlett, R.; Price, S. Intensive Care Management of the Cardiogenic Shock Patient. US Cardiology Review 2022;16:e20. 2022. DOI: 10.15420/usc.2021.23.
(42) Konstantinides, S. V.; Meyer, G.; Becattini, C.; Bueno, H.; Geersing, G.-J.; Harjola, V.-P.; Huisman, M. V.; Humbert, M.; Jennings, C. S.; Jiménez, D.; et al. 2019 ESC Guidelines for the diagnosis and management of acute pulmonary embolism developed in collaboration with the European Respiratory Society (ERS). The Task Force for the diagnosis and management of acute pulmonary embolism of the European Society of Cardiology (ESC) 2019, 1901647. DOI: 10.1183/13993003.01647-2019.
(43) Ohle, R.; Yan, J. W.; Yadav, K.; Cournoyer, A.; Savage, D. W.; Jetty, P.; Atoui, R.; Bittira, B.; Wilson, B.; Gupta, A.; et al. Diagnosing acute aortic syndrome: a Canadian clinical practice guideline. Cmaj 2020, 192 (29), E832-e843. DOI: 10.1503/cmaj.200021 From NLM.
(44) Vagnarelli, F.; Corsini, A.; Lorenzini, M.; Pacini, D.; Ferlito, M.; Bacchi Reggiani, L.; Longhi, S.; Nanni, S.; Norscini, G.; Cinti, L.; et al. Acute heart failure in patients with acute aortic syndrome: pathophysiology and clinical-prognostic implications. Eur J Heart Fail 2015, 17 (9), 917-924. DOI: 10.1002/ejhf.325 From NLM.
(45) Fukui, T. Management of acute aortic dissection and thoracic aortic rupture. Journal of Intensive Care 2018, 6 (1), 15. DOI: 10.1186/s40560-018-0287-7.
(46) Stout, K. K.; Verrier, E. D. Acute Valvular Regurgitation. Circulation 2009, 119 (25), 3232-3241. DOI: doi:10.1161/CIRCULATIONAHA.108.782292.
(47) Habimana, R.; Choi, I.; Cho, H. J.; Kim, D.; Lee, K.; Jeong, I. Sepsis-induced cardiac dysfunction: a review of pathophysiology. Acute Crit Care 2020, 35 (2), 57-66. DOI: 10.4266/acc.2020.00248 From NLM.
(48) L’Heureux, M.; Sternberg, M.; Brath, L.; Turlington, J.; Kashiouris, M. G. Sepsis-Induced Cardiomyopathy: a Comprehensive Review. Curr Cardiol Rep 2020, 22 (5), 35. DOI: 10.1007/s11886-020-01277-2 From NLM.
(49) Kakihana, Y.; Ito, T.; Nakahara, M.; Yamaguchi, K.; Yasuda, T. Sepsis-induced myocardial dysfunction: pathophysiology and management. Journal of Intensive Care 2016, 4 (1), 22. DOI: 10.1186/s40560-016-0148-1.
(50) Veronese, G.; Ammirati, E.; Cipriani, M.; Frigerio, M. Fulminant myocarditis: Characteristics, treatment, and outcomes. Anatol J Cardiol 2018, 19 (4), 279-286. DOI: 10.14744/AnatolJCardiol.2017.8170 From NLM.
(51) Gottlieb, M.; Bridwell, R.; Petrak, V.; Long, B. Diagnosis and Management of Myocarditis: An Evidence-Based Review for the Emergency Medicine Clinician. J Emerg Med 2021, 61 (3), 222-233. DOI: 10.1016/j.jemermed.2021.03.029 From NLM.
(52) Baud, F. J.; Megarbane, B.; Deye, N.; Leprince, P. Clinical review: Aggressive management and extracorporeal support for drug-induced cardiotoxicity. Critical Care 2007, 11 (2), 207. DOI: 10.1186/cc5700.
(53) Woodward, C.; Pourmand, A.; Mazer-Amirshahi, M. High dose insulin therapy, an evidence based approach to beta blocker/calcium channel blocker toxicity. Daru 2014, 22 (1), 36. DOI: 10.1186/2008-2231-22-36 From NLM.
(54) St-Onge, M.; Anseeuw, K.; Cantrell, F. L.; Gilchrist, I. C.; Hantson, P.; Bailey, B.; Lavergne, V.; Gosselin, S.; Kerns, W., II; Laliberté, M.; et al. Experts Consensus Recommendations for the Management of Calcium Channel Blocker Poisoning in Adults. Critical Care Medicine 2017, 45 (3).
(55) Skoog, C. A.; Engebretsen, K. M. Are vasopressors useful in toxin-induced cardiogenic shock? Clinical Toxicology 2017, 55 (4), 285-304. DOI: 10.1080/15563650.2017.1284329.
(56) Bouchard, J.; Shepherd, G.; Hoffman, R. S.; Gosselin, S.; Roberts, D. M.; Li, Y.; Nolin, T. D.; Lavergne, V.; Ghannoum, M.; Bouchard, J.; et al. Extracorporeal treatment for poisoning to beta-adrenergic antagonists: systematic review and recommendations from the EXTRIP workgroup. Critical Care 2021, 25 (1), 201. DOI: 10.1186/s13054-021-03585-7.
(57) Body, R.; Bartram, T.; Azam, F.; Mackway-Jones, K. Guidelines in Emergency Medicine Network (GEMNet): guideline for the management of tricyclic antidepressant overdose. Emergency Medicine Journal 2011, 28 (4), 347-368. DOI: 10.1136/emj.2010.091553.
(58) Bussienne, F.; Betello, M. Cardiogenic Shock Related to Carbon Monoxide Poisoning. Case Reports in Acute Medicine 2021, 4 (1), 32-35. DOI: 10.1159/000514303.
(59) Orliaguet, G.; Ferjani, M.; Riou, B. The Heart in Blunt Trauma. Anesthesiology 2001, 95 (2), 544-548. DOI: 10.1097/00000542-200108000-00041 (acccessed 12/25/2022).
(60) Maury, P.; Mansourati, J.; Fauchier, L.; Waintraub, X.; Boveda, S.; Sacher, F. Management of sustained arrhythmias for patients with cardiogenic shock in intensive cardiac care units. Archives of Cardiovascular Diseases 2019, 112 (12), 781-791. DOI: https://doi.org/10.1016/j.acvd.2019.10.002.
(61) Domienik-Karłowicz, J.; Kupczyńska, K.; Michalski, B.; Kapłon-Cieślicka, A.; Darocha, S.; Dobrowolski, P.; Wybraniec, M.; Wańha, W.; Jaguszewski, M. Fourth universal definition of myocardial infarction. Selected messages from the European Society of Cardiology document and lessons learned from the new guidelines on ST-segment elevation myocardial infarction and non-ST-segment elevation-acute coronary syndrome. Cardiol J 2021, 28 (2), 195-201. DOI: 10.5603/CJ.a2021.0036 From NLM.
(62) Forsberg, J.; Bedard, E.; Mahmoud, S. H. Bioavailability of Orally Administered Drugs in Critically Ill Patients. Journal of Pharmacy Practice 0 (0), 08971900221100205. DOI: 10.1177/08971900221100205.
(63) Boskovski, M. T.; Gleason, T. G. Current Therapeutic Options in Aortic Stenosis. Circulation Research 2021, 128 (9), 1398-1417. DOI: doi:10.1161/CIRCRESAHA.121.318040.
(64) Caetano, F.; Almeida, I.; Seca, L.; Botelho, A.; Mota, P.; Leitão Marques, A. Severe aortic stenosis and cardiogenic shock: A therapeutic challenge. Revista Portuguesa de Cardiologia (English edition) 2013, 32 (9), 701-706, 10.1016/j.repce.2013.10.012. DOI: 10.1016/j.repce.2013.10.012.
(65) Augustin, K. CV-EMCrit 327 – Acute Valve Disasters Part 2 – Management of Critical Aortic Stenosis. In EMCrit RACC, Connor-Schuler, R., Ed.; 2022; Vol. 2022.
(66) Jentzer, J. C.; Ternus, B.; Eleid, M.; Rihal, C. Structural Heart Disease Emergencies. J Intensive Care Med 2021, 36 (9), 975-988. DOI: 10.1177/0885066620918776 From NLM.
(67) Tandar, A.; Drakos, S. G. Cardiogenic shock in aortic stenosis patients: Balancing between complexity and simplicity. Hellenic Journal of Cardiology 2019, 60 (3), 182-184. DOI: https://doi.org/10.1016/j.hjc.2019.06.002.
(68) Toutouzas, P.; Koidakis, A.; Velimezis, A.; Avgoustakis, D. Mechanism of diastolic rumble and presystolic murmur in mitral stenosis. Br Heart J 1974, 36 (11), 1096-1105. DOI: 10.1136/hrt.36.11.1096 From NLM.
(69) Akodad, M.; Schurtz, G.; Adda, J.; Leclercq, F.; Roubille, F. Management of valvulopathies with acute severe heart failure and cardiogenic shock. Archives of Cardiovascular Diseases 2019, 112 (12), 773-780. DOI: https://doi.org/10.1016/j.acvd.2019.06.009.
(70) Wigle, E. D.; Rakowski, H.; Kimball, B. P.; Williams, W. G. Hypertrophic Cardiomyopathy. Circulation 1995, 92 (7), 1680-1692. DOI: doi:10.1161/01.CIR.92.7.1680.
(71) Evans, J. S.; Huang, S. J.; McLean, A. S.; Nalos, M. Left ventricular outflow tract obstruction-be prepared! Anaesth Intensive Care 2017, 45 (1), 12-20. DOI: 10.1177/0310057×1704500103 From NLM.
(72) Di Vece, D.; Silverio, A.; Bellino, M.; Galasso, G.; Vecchione, C.; La Canna, G.; Citro, R. Dynamic Left Intraventricular Obstruction Phenotype in Takotsubo Syndrome. J Clin Med 2021, 10 (15). DOI: 10.3390/jcm10153235 From NLM.
(73) Maiden, M.; Payne, J.; Shu, A. Cardiogenic Shock Due to Atrial Myxoma With Mitral Valve Involvement. Cureus 2022, 14 (8), e28520. DOI: 10.7759/cureus.28520 From NLM.
(74) Kearns, M. J.; Walley, K. R. Tamponade: Hemodynamic and Echocardiographic Diagnosis. Chest 2018, 153 (5), 1266-1275. DOI: 10.1016/j.chest.2017.11.003 From NLM.
(75) Alerhand, S.; Carter, J. M. What echocardiographic findings suggest a pericardial effusion is causing tamponade? Am J Emerg Med 2019, 37 (2), 321-326. DOI: 10.1016/j.ajem.2018.11.004 From NLM.

One thought on “Cardiogenic Shock: Emergency Department-Focused Management”