
Author: David Cisewski, MD (@PainProfiles – EM Resident Physician, Icahn School of Medicine at Mount Sinai) // Edited by: Manpreet Singh, MD (@MPrizzleER); Alex Koyfman, MD (@EMHighAK); and Brit Long, MD (@long_brit)

Last week marked the release of the updated American Society of Hematology (ASH) ASH Draft Recommendations Sickle Cell Disease-Related Pain. These guidelines have been released as a draft and are currently open for public commentary through May 13, 2019. This EMDocs article addresses each of the draft recommendations listed on behalf of ASH as related to the ED-setting. Analgesic options such as cognitive behavioral therapy, acupuncture, massage, yoga, TENS were also addressed as draft recommendations though not covered in this review due the limitation of their use in the emergency setting – please see the above link for a full list of 10 recommendations).
Case
A 22-year-old male with known sickle cell disease presents to the emergency triage requesting opioids for what he feels is a sickle cell vaso-occlusive crisis. The patient indicates he missed his pain clinic appointment and has run out of his home Percocet prescription three days ago. Since then he has noted progressively worsening pain in his lower back and lower extremities that he believes was precipitated by an upper respiratory infection. He Indicates the pain is currently 10/10. He denies fever, chest pain, shortness of breath, vision changes, headache, or abdominal pain. The nursing staff is suspicious of drug-seeking activity as this is the third time in six weeks the patient has presented with similar complaints, each time specifically requesting ‘Dilaudid’. After initial vital signs are recorded to be within normal limits – blood pressure 128/88, heart rate 81, temp 37.1C, RR 13, O2 98% – the patient is triaged back to the waiting room to wait for an emergency department (ED) bed for further medical evaluation. Due to a backed-up ED, the patient is not seen by a provider until two hours after arrival. IV hydromorphone is ordered but due to several failed attempts to obtain IV access, treatment is further delayed. An emergency resident obtains ultrasound-guided IV access, and three hours after arrival to the ED, the patient receives his first analgesic treatment.
Sickle Cell Vaso-occlusive Crises
First described by Dr. James Herrick in 1910, sickle cell disease (SCD) is estimated to effect over 100,000 individuals, making it the most common genetic disease in the United States [1]. Although SCD can affect any system throughout the body, vaso-occlusive crises (VOC) remain one of the most common and debilitating pathologies among individual presenting to the ED [2]. The purpose of this article is to shed light on the myths and facts of VOC pain management in the ED setting and to share a novel approach to VOC pain management, which can be used among SCD patients who present during these debilitating events (highlighting the ASH Draft Recommendations Sickle Cell Disease-Related Pain as they relate to the ED setting).
First off… how does a VOC occur?
As opposed to normal hemoglobin (HbA) which can transition quickly between an oxygenated and deoxygenated form, sickle cell hemoglobin (HbSC) forms rigid polymers when deoxygenated creating the characteristic “sickle shape”. These sickled polymers back up and engorge post-capillary venules and bone marrow. Though common belief was that these sickled cells caused VOC pain, it is now believed that it is the precipitation of a cascade of inflammatory responses that lead to the excruciatingly painful presentation referred to as a ‘sickle cell crisis’. Patients in an acute crisis are most likely to present with pain along marrow-containing areas such as the lower back and lower extremities (most common), arms, shoulders, sternum, and clavicle. SCD patients are acutely aware of their typical symptomology, so it’s important to ask whether the current symptoms are consistent or different from prior episodes to assess for an alternative source of pain.
But this guy is always coming in demanding Dilaudid for his VOC pain… how do I know this isn’t just drug-seeking behavior?
Years of repeated VOC’s can result in substantial analgesic tolerance,and higher doses of analgesics are required to alleviate the pain during these events. There is a subset of patients for which high-frequency, high-dose analgesic regimens may be ineffective, but this should not be misinterpreted as drug-seeking behavior; do not discount pain as opioid-seeking behavior until discussing the case with the pain team or hematologist. The true incidence of opiate addiction among SCD patients is less than 5%, and even high ED utilizers do not use opioids more frequently than other sickle cell disease patients in the community [3].
But the patient’s vital signs are normal so they can’t be having a vaso-occlusive crisis, right?
Yes, they can. In all patients presenting to the ED – regardless of underlying pathology – research has shown vital signs are an inherently unreliable predictor of overall pain and should not be used to assess degree of severity pain [4, 5]. If a patient presents with the symptoms of a VOC, it is important that we address the pain until proven otherwise.
How do I know this isn’t just chronic pain associated with recurrent VOC episodes?
We’ve heard a lot about the need to limit opioid use among patients presenting with chronic pain conditions due to a lack of efficacy compared to non-opioid alternatives(ie, risks outweigh benefits). Despite living with low levels of chronic pain that many SCD patients become accustomed to, vaso-occlusive crises, no matter how recurrent, are not considered ‘chronic pain’. All VOC’s are considered a form of acute pain, regardless of how long the patient has been having VOC, and should be treat as such (as opposed to chronic pain management which would focus on an opioid-free regimen).
Great – so if a patient says they’re having an acute crisis, this saves me time in the workup, right?
Although VOC is the most common reasons for SCD patients to present to the ED, it is important to note VOC is a diagnosis of exclusion [2, 6]. EM providers must avoid anchoring on the ‘common presentation’ and ensure other forms of life-threatening SCD pathology have been excluded (see table 1 for a list of items to consider in the differential).
What are the signs and symptoms I should be evaluating for to eliminate alternative SCD pathology in a VOC patient?
All patients, despite the consistency of the symptomology with prior VOC presentations, warrant a full physical exam. Red flags include abnormal vital signs (febrile, tachycardia, tachypneic, hypotensive), shortness of breath, chest pain, headache, confusion, altered mental status, seizure activity, priapism, and patients who present during pregnancy. Additionally, red or swollen joints should raise concern for cellulitis, osteomyelitis, or avascular necrosis instead of VOC pain (see table 1). Don’t forget that these patients are equally likely to present with core pathology as well (e.g. chest pain radiating to the shoulders with nausea and vomiting is ACS until proven otherwise).

Table 1: Differential diagnosis for a sickle cell vaso-occlusive crisis. Sickle cell patients have an increased susceptibility to certain disease pathologies that should be considered depending on the pain type/location.
So if the physical exam is otherwise normal and the patient says this is a VOC, do I even need lab tests?
Lab tests can assist in assessing the severity of the underlying crisis, the body’s compensatory response to the crisis, and to direct the pretest probability of an infectious source. Be cautious about worsening anemia (compare to patient’s baseline hemoglobin), thrombocytopenia (increased bleeding risk), elevated lactate dehydrogenase (evidence of RBC destruction), and a low reticulocyte count (indication of an inadequate bone marrow response or an aplastic crisis). Additionally, bandemia with leukocytosis at any white blood cell count should raise concern for an underlying bacterial infection.
Okay, I need to do a really good physical exam to assess the pain…. I can hold off on treating the pain until after a full physical examination and workup have been performed to eliminate alternative sources of pain, right?
Treating the pain will not limit your physical exam. The first rule in all SCD VOC patients is to perform a rapid assessment and initiate an analgesic regimen within one hour of ED arrival (draft recommendation 1). Aggressive VOC pain management has been shown to prevent the sequelae of unrelieved pain, decrease duration of pain, and decrease hospital admission rate[7, 8]. Always assess for an individualized VOC pain regimen, but if a plan does not exist, have an anticipated algorithm including oral, nebulized, subcutaneous, and intravenous analgesics to initiate when these patients present (see below). Additionally,frequent reassessments every 30-60 minutes are recommended to optimize pain management (draft recommendation 1).
But these SCD patients are always hard sticks – what if the nurse can’t place a line?
Consider early initiation of an analgesic bridge with nebulized, subcutaneous (SC), or intranasal (IN) formulations (see below) if your institution provides. Consider initiating a program at your institution that allows the nursing staff in triage to initiate these medication to reduce mean time to first-dose analgesia if the facility allows [9].

Table 2: List of commonly used intranasal analgesics used in the (ED) setting. Among SCD patient in which IV access cannot be obtained, consider intranasal analgesic formulations as a bridge to pain management.
Okay, I have IV access but no individualized pain management plan for this patient… where should I start?
Opioids are still considered a first-line regimen during a VOC. However, typical pain management regimens are not usually effective in treating a vaso-occlusive crisis, and higher doses are often required to achieve pain relief. Consider longer acting opioids such as morphine and hydromorphone. Although a standard dosing of 0.1 mg/kg IV morphine is used for acute pain presentations in the ED, consider 0.15 mg/kg IV as a standard starting dose among opioid-tolerant SCD patients. Similarly, consider 0.01-0.02 mg/kg IV hydromorphone as an equianalgesic opioid alternative (see figure 1 for a novel approach to VOC pain management in the ED setting).
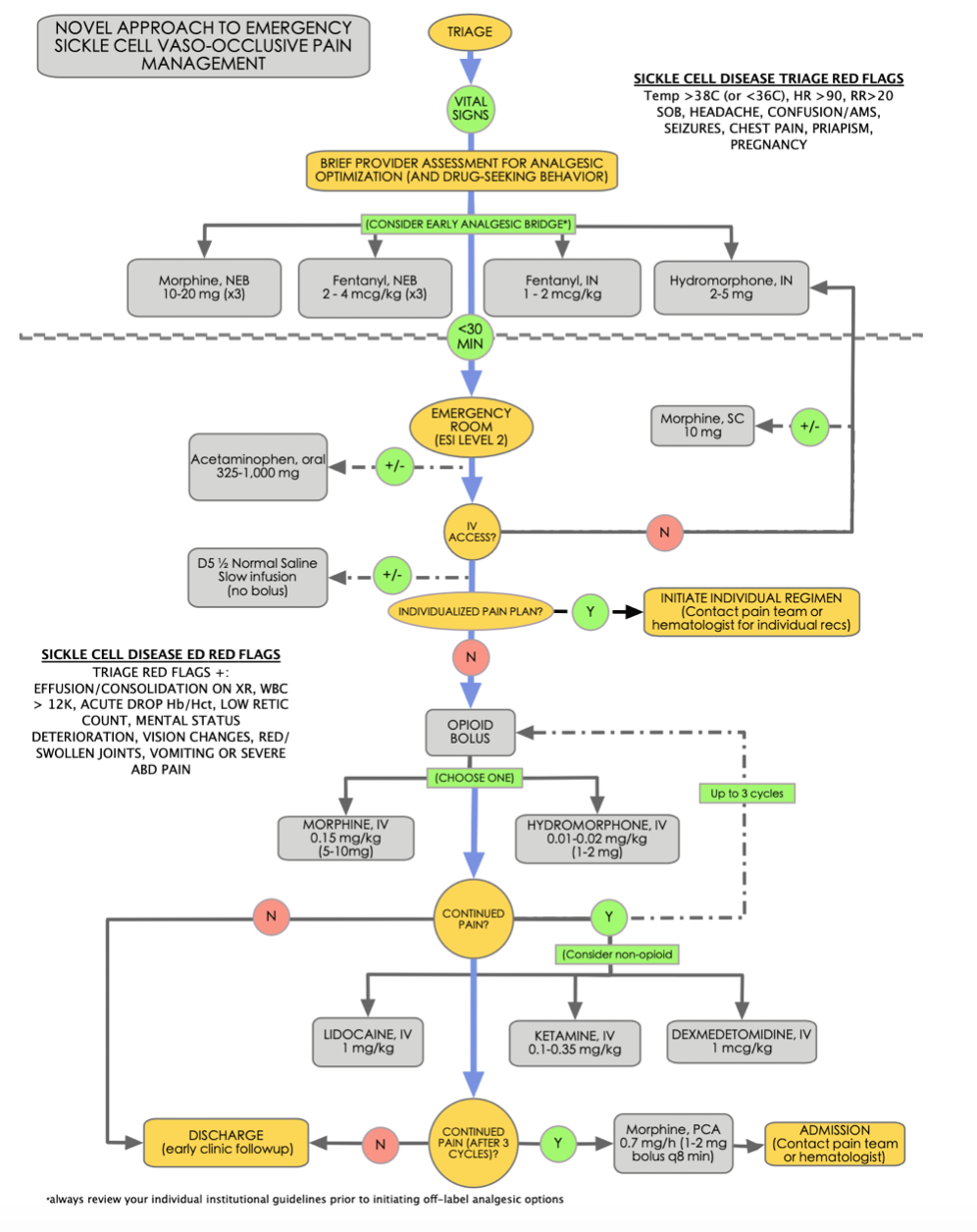
Figure 1: Novel approach to emergency sickle cell vaso-occlusive pain management in the ED. This approach focuses on early assessment, expedited time to first-dose, and rapid reassessments and re-dosing analgesia as needed while always considered the red flags of alternative pain pathology.
Do all patients require Dilaudid for pain?
Despite efforts to minimize opioid use in the ED, doing so among the SCD patient population will inevitably lead to poor pain control and unnecessary patient suffering. Optimization of VOC pain should be a priority (draft recommendation 9). However, novel approaches to opioid weaning are being developed by hematologists and sickle cell clinics, which means many patients may not need or want maximum opioids despite a crisis. Always involve the patient in the analgesic plan and allow shared decision making after discussing the risks and benefits of all available options as well as the risk of chronic non-SCD pain associated with long-term opioid use. Additionally, always check the EMR and previous clinic records to assess for an individualized pain regimen that may be tailored to the patient’s specific needs/desire for pain management (draft recommendation 9).
Is there anything I can give besides opioids?
Consider non-opioid adjuncts as a means of complimenting the opioid analgesic regimen (draft recommendation 1). Ketamine has been shown to lower overall opioid use among admitted SCD patients [10]. A sub-anesthetic dose of ketamine is recommended as a non-opioid alternative [0.1-0.35mg/kg ideal body weight (IBW) max; use actual body weight (ABW) if patient below IBW)] (draft recommendation 2). Dexmedetomidine and lidocaine are alternative analgesics that are increasing in popularity in the ED setting, though further data is needed to understand the safety and efficacy of these analgesics among VOC patients in the ED setting. Fibromyalgia is considered the disease entity most closely aligned with chronic pain among SCD patients, and tricyclic antidepressants (amitriptyline) may be considered as an analgesic adjunct (draft recommendation 6).
Can I give NSAIDS as a non-opioid adjunct?
It is safe to assume that all patients with SCD have at least a mild degree of renal dysfunction due to low-level renal microinfarcts, and that this may not be accurately reflected by creatinine levels. NSAIDs should be limited considering the availability of alternative analgesic options. If necessary, a short-course of no more than 5-7 days of NSAIDs is recommended for acute pain management (draft recommendation 2).
I’ve heard dehydration can cause sickling as well… should I start a liter of fluids?
Never bolus IVF in SCD patients. Despite dehydration being a common precipitant of VOC’s, rehydration can result in atelectasis, pulmonary edema, and acute chest syndrome. Normal saline may also promote hyperchloremic metabolic acidosis, further precipitating sickling [6]. Additionally, recent research has shown normal saline boluses have been associated with worse pain control and increased rates of admission among patients presenting with acute VOC pain [11, 12]. If concerned for dehydration, start a D5 ½ normal saline infusion at maintenance rate.
If deoxygenation causes sickling… maybe they just need a little oxygen instead of opioids… should I start them on a nasal cannula to bump up their hemoglobin saturation?
Although there are no specific guidelines for administering supplemental oxygen, there is a theoretical risk that oxygen may inhibit the release of hypoxia-induced transcription factors (HIF) necessary for erythropoietin production, further delaying essential hematopoiesis among sickling patients [13]. At our center, patients with VOP are given supplemental oxygen only if saturation falls below 95% and down-titrated as early as possible. All patients being admitted should receive an incentive spirometer which has been shown to prevent atelectasis and acute chest syndrome development [6, 14].
The patient continues to have pain despite 3 rounds of hydromorphone, acetaminophen, and a bolus of ketamine, and requires admission… now what?
Once patient admitted, your job is not done – if possible, initiate an analgesic PCA to bridge patient to inpatient management. If available, initiate a discussion with the inpatient pain management team to optimize the SCD pain management regimen (draft recommendation 9). Research has shown that around-the-clock analgesic infusions over the first 24 hours following admission (this can include ED transition) were more effective for managing VOCs than patient-requested infusions of analgesics and resulted in a higher discharge rate within 72 hours of admission (Udezue, 2007). Morphine PCA among VOC patients has shown to reduce opioid consumption and improve the side effect profile compared to continuous infusion [15], a shorter length of ED stay [16], and a decrease in opioid bolus frequency with no observed difference in pain reduction [17]. The use of a continuous opioid infusion in conjunction with on-demand dosing or scheduled intermittent dosing is per provider discretion,as there is no direct evidence to support either intervention (draft recommendation 5). For institutions without PCA capability, ensure frequent patient pain reassessments are completed every 30-60 minutes until patient is dispositioned to the inpatient unit.
The Upshot
Vaso-occlusive crisis pain is a common, debilitating pathology among sickle cell patients and should not be misinterpreted as drug-seeking behavior. Early recognition and initiation of an analgesic regimen is essential to limiting further sequelae and hospital admission rates/length of stay. Do not rely on abnormal vital signs (or absence of) to guide your management. Oral, intranasal, subcutaneous, nebulized, and intravenous routes of administration should all be considered. Remember that typical pain management regimens are not usually effective in treating a vaso-occlusive crisis. Limit NSAIDS and avoid fluid boluses during acute pain management. Limit oxygen to patients with O2 saturation < 95%. If admission is required, ensure a smooth transition of care that involves a continued analgesic regimen and involvement of the pain management or hematologic consultation service.

Special thanks to Dr. Jeffrey Glassberg for his assistance and contributions to this article. Dr. Glassberg is an Associate Professor of Emergency Medicine, Hematology and Medical Oncology and Associate Director of the Comprehensive Program for Sickle Cell Disease at Mount Sinai School of Medicine.
From Dr. Katelyn Hanson and Hanson’s Anatomy:

Further Reading
- NIH Evidence-Based Management of Sickle Cell Disease
- STAT News – ‘Every time it’s a battle’: In excruciating pain, sickle cell patients are shunted aside
- Interested in becoming an advocate for these practices? Check out the ACEPEmergency Department Sickle Cell Care Coalition
References:
- CDC. Data & Statistics on Sickle Cell Disease. 2017 August 9, 2017; Available from: https://www.cdc.gov/NCBDDD/sicklecell/data.html.
- Smith, W.R., et al., Daily assessment of pain in adults with sickle cell disease.Ann Intern Med, 2008. 148(2): p. 94-101.
- Aisiku, I.P., et al., Comparisons of high versus low emergency department utilizers in sickle cell disease.Ann Emerg Med, 2009. 53(5): p. 587-93.
- Dayoub, E.J. and A.B. Jena, Does Pain Lead to Tachycardia? Revisiting the Association Between Self-reported Pain and Heart Rate in a National Sample of Urgent Emergency Department Visits.Mayo Clin Proc, 2015. 90(8): p. 1165-6.
- Ernst, A.A., et al., Blood pressure in acute vaso-occlusive crises of sickle cell disease.South Med J, 2000. 93(6): p. 590-2.
- Glassberg, J., Evidence-based management of sickle cell disease in the emergency department.Emerg Med Pract, 2011. 13(8): p. 1-20; quiz 20.
- Benjamin, L.J., G.I. Swinson, and R.L. Nagel, Sickle cell anemia day hospital: an approach for the management of uncomplicated painful crises.Blood, 2000. 95(4): p. 1130-6.
- Tanabe, P., et al., A randomized controlled trial comparing two vaso-occlusive episode (VOE) protocols in sickle cell disease (SCD).Am J Hematol, 2018. 93(2): p. 159-168.
- Kavanagh, P.L., et al., Improving the Management of Vaso-Occlusive Episodes in the Pediatric Emergency Department.Pediatrics, 2015. 136(4): p. e1016-25.
- Tawfic, Q.A., A.S. Faris, and R. Kausalya, The role of a low-dose ketamine-midazolam regimen in the management of severe painful crisis in patients with sickle cell disease.J Pain Symptom Manage, 2014. 47(2): p. 334-40.
- Carden, M.A., et al., Variations in pediatric emergency medicine physician practices for intravenous fluid management in children with sickle cell disease and vaso-occlusive pain: A single institution experience.Pediatr Blood Cancer, 2018. 65(1).
- Carden, M.A., et al., Normal Saline Bolus Use in Pediatric Emergency Departments is Associated with Worse Pain Control in Children with Sickle Cell Anemia and Vaso-occlusive Pain.Am J Hematol, 2019.
- Frede, S., et al., Oxygen-regulated expression of the erythropoietin gene in the human renal cell line REPC.Blood, 2011. 117(18): p. 4905-14.
- Bellet, P.S., et al., Incentive spirometry to prevent acute pulmonary complications in sickle cell diseases.N Engl J Med, 1995. 333(11): p. 699-703.
- van Beers, E.J., et al., Patient-controlled analgesia versus continuous infusion of morphine during vaso-occlusive crisis in sickle cell disease, a randomized controlled trial.Am J Hematol, 2007. 82(11): p. 955-60.
- Gonzalez, E.R., et al., Intermittent injection vs patient-controlled analgesia for sickle cell crisis pain. Comparison in patients in the emergency department.Arch Intern Med, 1991. 151(7): p. 1373-8.
- Santos, J., et al., Patient Controlled Analgesia for Adults with Sickle Cell Disease Awaiting Admission from the Emergency Department.Pain Res Manag, 2016. 2016: p. 3218186.





1 thought on “ED Management of Sickle Cell Vaso-occlusive Crises: Myths, Facts, and A Novel Approach to Acute Pain Management”
Pingback: April FOAMed - FRCEM Success