Authors: Baruch Berzon, MD, and Richard Sinert, MD (Kings County Hospital-SUNY Downstate) // Edited by: Alex Koyfman, MD (@EMHighAK), Brit Long, MD (@long_brit, EM Resident at SAUSHEC, USAF)
Case Presentation
A 32 year-old male presents via EMS with complaints of “fluttering in my chest” and lightheadedness for an hour. The paramedics find a diaphoretic young man in mild distress with heart rate of 220 and blood pressure of 110/69. The 3-lead ECG shows a wide-complex tachycardia at a rate of 220 BPM. They obtain IV access and perform a 12-lead ECG. They transmit the ECG and call you for consultation:
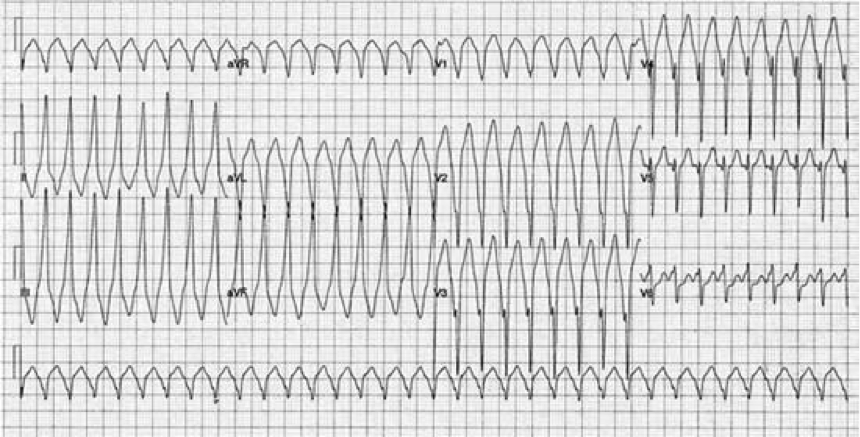
Clinical questions
1. Interpret the above 12 lead ECG.
2. Should they attempt to treat this chemically or electrically? If chemically, what is the drug of choice?
3. They converted the patient and get the following repeat ECG:
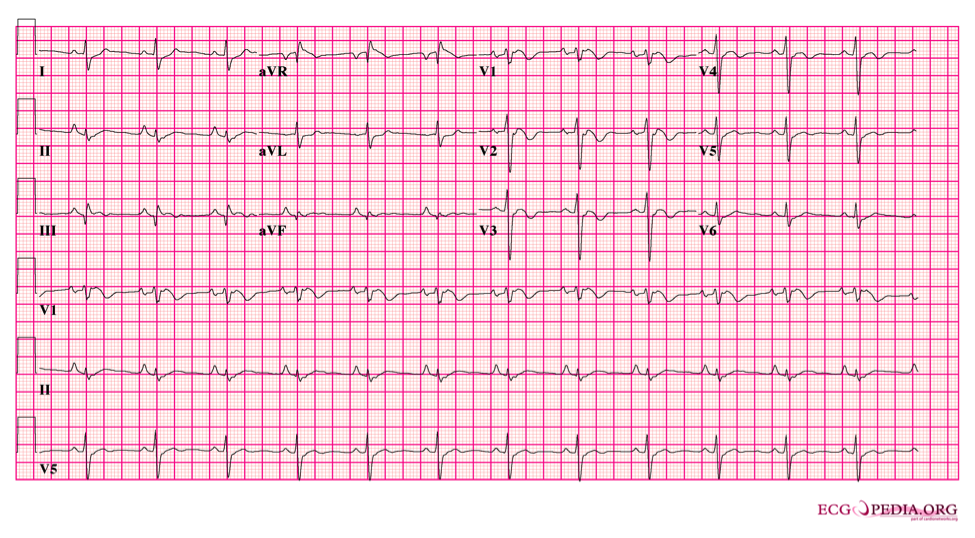 4. Now that the patient is in your ED, his vital signs are normal. In addition to routine lab testing and cardiology consultation for ventricular tachycardia, what else would you do for the care of this patient?
4. Now that the patient is in your ED, his vital signs are normal. In addition to routine lab testing and cardiology consultation for ventricular tachycardia, what else would you do for the care of this patient?
Overview
Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy (ARVD/C) is one of the most arrhythmogenic forms of inherited cardiomyopathy and a cause of sudden cardiac death (SCD) in the young.1 ARVD is the most common cause of sudden cardiac death in young athletes after hypertrophic cardiomyopathy.4
Pathophysiology – ARVD/C is inherited in an autosomal dominant manner, but because it has incomplete penetrance and variable expressivity, affected individuals have varying clinical courses. The eight genes involved encode desmosome proteins that act as a structural connection between cardiac cells and are also involved in calcium hemostasis and cell-to-cell signaling. When cell-to-cell binding is affected, cells undergo apoptosis and are replaced by fat cells and fibrous tissue. The disease is characterized by fibro-fatty replacement of predominantly the right ventricle (RV), which predisposes patients to life-threatening ventricular arrhythmias as well as progressive mechanical ventricular dysfunction. As the infiltration progresses, it eventually can lead to LV involvement and failure. The fatty or fibrofatty infiltration occurs at the epicardium of the RV and often involves the “triangle of dysplasia” which is bordered by the apex, infundibulum, and the inferior aspect of the RV but can progress to include the myocardium as well as infiltration of the septum and LV.
Prevalence – ARVD/C has an estimated prevalence of 1/1000 to 1/5000 Caucasian individuals, although some studies report that the real prevalence could be higher due to under-recognition.2 In some populations, it may be even more prevalent (one Finish study finds 1/200).3
Clinical presentation – ARVD/C usually presents in the 3rd to 5th decades of life, and patients may experience syncope, ventricular dysrhythmias ranging from PVCs to VT to VF, heart failure, and/or sudden cardiac death.
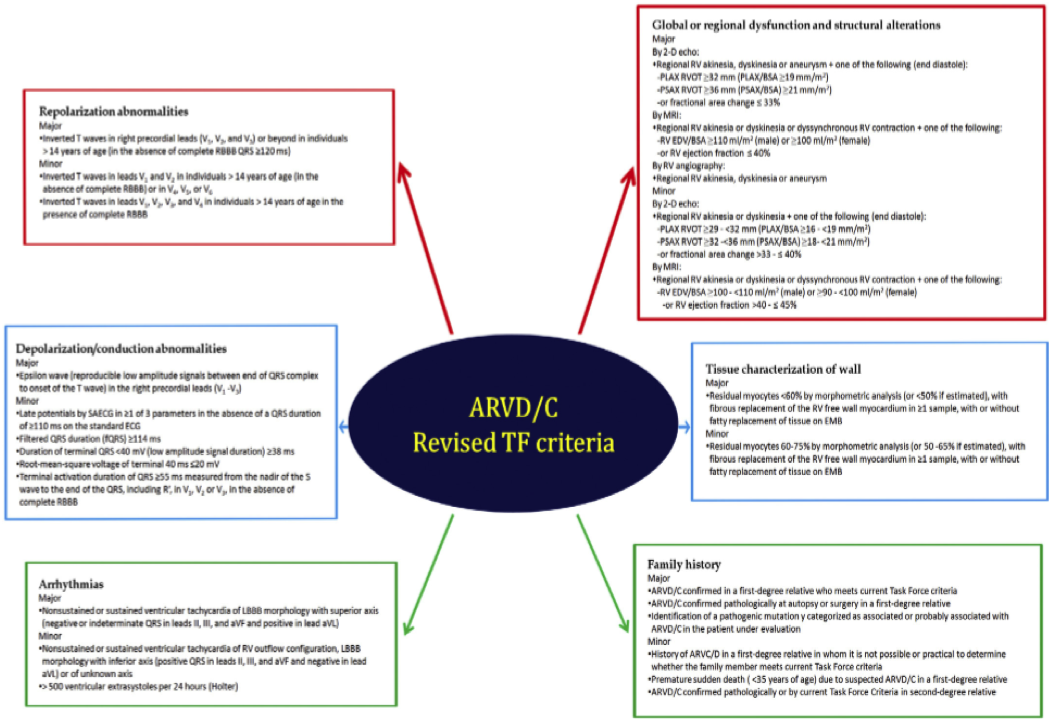
Diagnosis – The diagnosis is complex for several reasons: the heterogeneity of presentation, the low yield of endomyocardial biopsy (EMB), and the different genetics involved. The diagnosis is therefore based on Task Force criteria in six different aspects of the disease. They were initially published in 1994, with revision and validation in 2010. They are summarized below:5
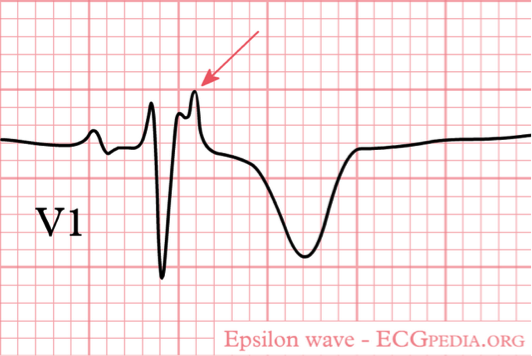
The classic ECG finding is an Epsilon wave, which is a low amplitude signal at the end of the QRS in V1-V3.
One can infer from the above that the testing required to make a definite diagnosis is not readily available in the ED. However a history of SCD in the family, previous unexplained syncopal episodes, and an ECG with characteristic findings should raise the clinical suspicion for ARVD/C and prompt further testing.
Management – Initial management should always include the ABCs. In unstable patients with ventricular arrhythmias, cardioversion is recommended. In stable patients, amiodarone remains the most effective empiric anti-arrhythmic for ventricular dysrhythmias (of note: procainamide has become agent of choice for many given higher rate of success).5 Checking electrolytes and evaluating for precipitating factors such as infection is warranted. Consultation with the cardiologist is paramount to further investigate for ARVD/C. If suspected, the patient will undergo 2D echo and /or cardiac MRI, signal average ECG, Holter monitoring, genetic analysis, and invasive studies such as RV Angio and Biopsy (EMB). Counseling other family members for early diagnosis and prevention of the disease is recommended, and relatives are further risk-stratified based on history and further testing.5
Long-term treatment includes anti-arrhythmic drugs for prevention of arrhythmias and implantation of an AICD. Catheter ablation with new 3D mapping has heterogeneous results with success rates ranging from 45-85%. In patients with extensive heart failure or drug-refractory VTs, heart transplantation is the last resort, but this occurs rarely.
In summary, ARVD/C is a heterogenic disease that presents in young patients with syncope, VTs, or sudden cardiac death. The epsilon wave might be the first clue for the emergency physician to the possibility of ARVD/C and should prompt a thorough history, exam, and appropriate diagnostic testing.
Extra! Extra! Read all about it–
Did you know that some ARVD/C has involvement of predominantly the LV?
Did you know that sometimes ARVD/C is recessive and involves a cutaneous phenotype? (e.g. Naxos disease or Carvajal syndrome)?
That there is a known link between ARVD/C and myocarditis that is still waiting for YOU to clearly define?
FOAMed and References
http://journal.frontiersin.org/article/10.3389/fphys.2012.00023/fullcomparison to RVOT
http://www.heartpearls.com/tag/epsilon-wave
http://lifeinthefastlane.com/ecg-library/basics/epsilon-wave/
http://www.af-ablation.org/?page_id=2034
http://www.revespcardiol.org/en/arrhythmia-and-right-heart-disease/articulo/13154581/ RVOT and Brugada looks at as well
- Arrhythmogenic right ventricular dysplasia/cardiomyopathy: Clinical challenges in a changing disease spectrum. te Riele A. et al; Trends in Cardiovascular Medicine, Volume 25, Issue 3, April 2015, Pages 191–198
- Cardiovascular causes of sudden death in young individuals including athletes. Basso C. et al; Cardiology Review, 7 (1999), pp. 127–135
- Population-prevalent desmosomal mutations predisposing to arrhythmogenic right ventricular cardiomyopathy. Lahtinen AM et al; Heart Rhythm.2011 Aug;8(8):1214-21
- Impact of new task force criteria in the diagnosis of arrhythmogenic right ventricular cardiomyopathy. Giuseppe F. et al; International Journal of Cardiology 171 (2014) 179–183
- Current and state of the art on the electrophysiologic characteristics and catheter ablation of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Chung F-P, et al; J Cardiol (2015)
- http://pubmed.org/pubmed/10577413
- http://pubmed.org/pubmed/25344388
- http://pubmed.org/pubmed/11146024





3 thoughts on “Arrhythmogenic Right Ventricular Dysplasia/Cardiomyopathy: EM Highlights”
Pingback: LITFL Review 185 | LITFL: Life in the Fast Lane Medical Blog
Pingback: AVRD/C | FOAM links
Pingback: Arrhythmogenic Right Ventricular Dysplasia (ARVD) • ECG Library