Author: Dan Troha, MD (Attending Physician, Carolinas Medical Center, Charlotte, North Carolina) // Edited by: Alex Koyfman, MD (@EMHighAK, EM Attending Physician, UT Southwestern Medical Center / Parkland Memorial Hospital) and Brit Long, MD (@long_brit, EM Chief Resident at SAUSHEC, USAF)
Case #1:
A 30-year-old female presents to the ED complaining of severe chest pain and dyspnea. She was diagnosed with a DVT 14 days ago but has been non-compliant with oral anticoagulation. Her temperature is 99.9 degrees Fahrenheit, heart rate 135, BP 74/49, respiratory rate 30, and oxygen saturation 85%. CTPA reveals large, bilateral pulmonary emboli at the bifurcation of her right and left main pulmonary arteries. What is the best treatment approach in this patient?
Case #2:
A 68-year-old male with a history of HTN presents to the ED complaining of acute onset dyspnea. On arrival, his temperature is 98.8 degrees Fahrenheit, heart rate 110, BP 105/70, respiratory rate 24, and oxygen saturation 87%. He appears dyspneic and anxious. The patient’s blood pressure drops to 69/43, and he begins to develop altered mental status. The patient is deemed too unstable to undergo CTPA. How do you best manage this patient’s undifferentiated shock?
Introduction
In the emergency department, we are responsible for identifying and treating life-threatening conditions. When a patient presents with acute onset dyspnea, chest pain, syncope, or hemoptysis, the diagnosis of pulmonary embolism (PE) often comes to mind; however, PE may escape prompt diagnosis when the clinical signs and symptoms are not as specific. Systemic arterial hypotension is a rare but life-threatening presentation of acute PE. The PIOPED II database suggests that only 8% of patients present with overt circulatory collapse.[i] Even in these patients, respiratory distress is present in 91%.
Massive (high risk) PE is defined as systolic BP less than 90 mm Hg or a systolic pressure drop greater than or equal to 40 mm Hg for greater than 15 minutes, not due to new-onset arrhythmia, hypovolemia, or sepsis.[ii] It should be noted that the word “massive” does not refer to clot burden, but refers to the presence of shock in the setting of acute PE. Massive PE carries a high mortality rate, likely somewhere around 30-50%.[iii] Because of this, aggressive measures must be taken to resuscitate and stabilize these patients.
Right Ventricular Failure
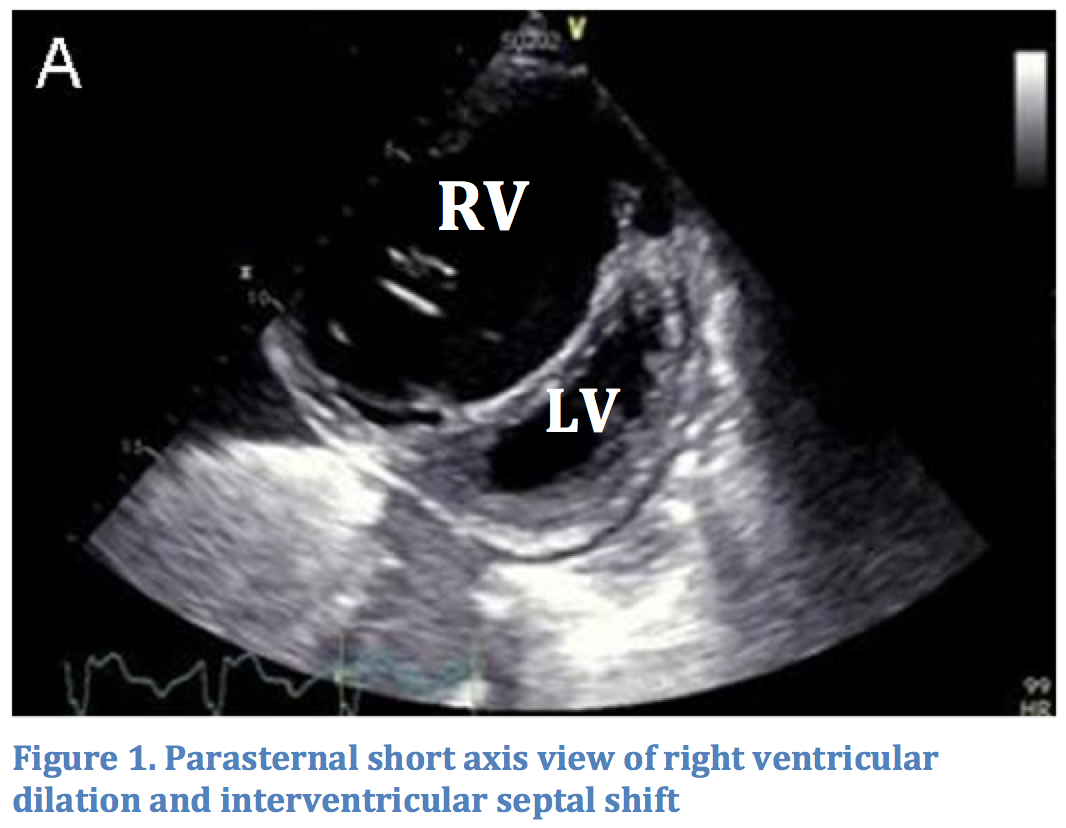
The circulatory collapse in massive PE starts with right ventricular (RV) dysfunction. Pulmonary arterial occlusion from thrombus increases pulmonary vascular resistance (PVR). The increase in pressure is transmitted to the RV. Unlike the left ventricle, the right ventricle is not capable of handling significant increases in PVR. Because they are connected in series, as right ventricular output falls, so does left ventricular output. Additionally, increased right ventricular pressures lead to RV dilation. This may cause interventricular septal shift, further decreasing diastolic filling of the left ventricle (see Figure 1).
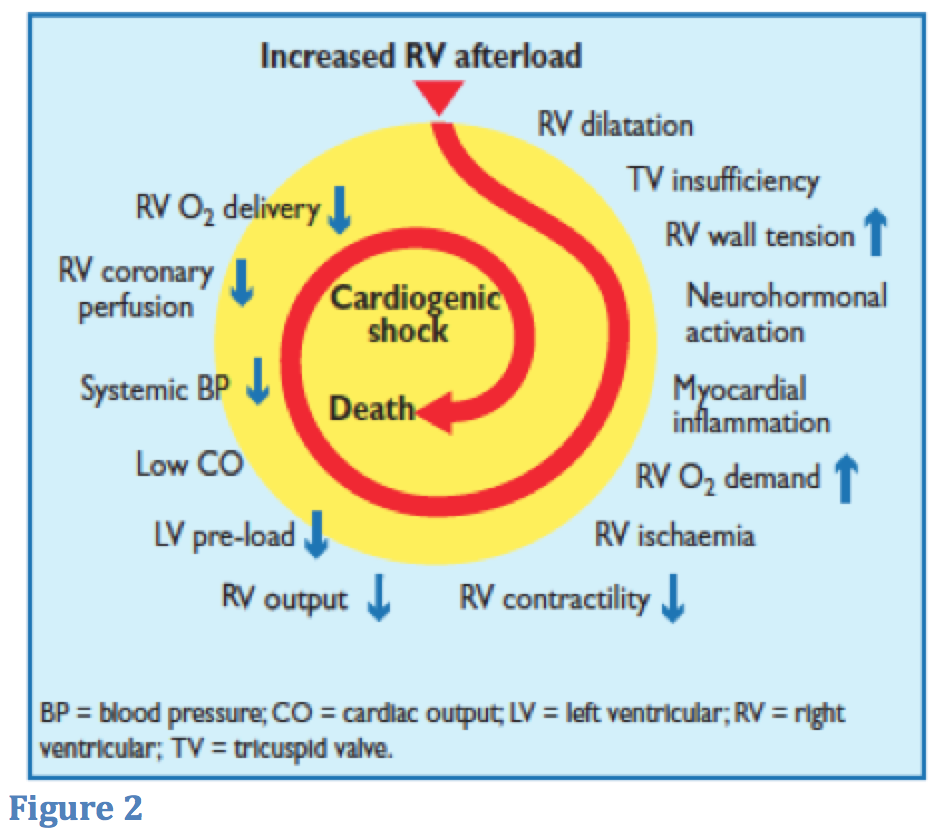
Both of these phenomena decrease cardiac output and ultimately, decrease systemic blood pressure. Drops in systemic blood pressure can worsen RV dysfunction because the RV is particularly prone to changes in coronary perfusion pressure. RV ischemia further propagates RV failure because of direct damage to myocytes. In Figure 2 below, obtained from the European Society of Cardiology 2014 Guidelines, it is clear that RV failure in the setting of acute PE can lead to rapid hemodynamic decompensation and death.
Methods to Decrease Pulmonary Vascular Resistance
In massive PE, immediate measures must be taken to decrease pulmonary vascular resistance. This, in turn, will decrease RV strain and hopefully reverse RV ischemia. The quickest way to decrease PVR is acute clot reduction. This is most easily performed by the administration of intravenous tissue plasminogen activator (tPA). Thrombolytic therapy leads to early hemodynamic improvement, but comes at a cost of major bleeding. The American Heart Association and CHEST guidelines recommend IV tPA for the treatment of massive PE in the absence of contraindications.[iv]
Catheter-directed therapy is an emerging option for acute clot reduction in PE. It may be considered for patients with persistent shock despite administration of systemic tPA. It is still considered investigational as a primary treatment modality.[v] The time required to mobilize an interventional team may result in further RV dysfunction and damage; therefore, systemic tPA is currently the recommended initial treatment modality.
If systemic tPA fails to improve hemodynamic instability, another therapeutic option is surgical embolectomy. Additionally, this intervention remains an option in situations where thrombolysis may be contraindicated. This procedure requires surgeons skilled at performing cardiothoracic interventions and necessitates cardiopulmonary bypass. In a pooled series of case reports, mortality after pulmonary embolectomy may be 30% or higher,[vi] so it must be reserved for situations where the potential benefits outweigh the risks of the procedure.
In addition to acute clot reduction, therapies aimed at pulmonary vasodilation may help with PVR reduction. Inhaled nitric oxide (iNO) has been proposed as a therapeutic intervention in acute PE based on its selective ability to dilate the pulmonary vasculature without significant effect on the systemic vasculature. With the exception of a Phase I clinical trial involving eight patients, no randomized controlled trials have compared iNO to placebo in the treatment of pulmonary embolism. Numerous case reports document the safety of iNO while reporting on its ability to improve oxygenation and hemodynamics in the setting of acute PE.[vii]
Airway Management
Pulmonary embolism can present with profound respiratory failure. The respiratory failure seen in PE is predominantly a result of hemodynamic disturbances.[viii] Even without significant hemodynamic abnormalities, distal emboli can create areas of alveolar hemorrhage resulting in the development of acute airspace disease – a phenomenon known as pulmonary infarction. Administration of supplemental oxygen improves ventilation-perfusion mismatch. Hypoxia can further increase PVR, so maintenance of normoxia is important.
Positive pressure ventilation via endotracheal intubation or non-invasive ventilation can have devastating effects on a patient with circulatory collapse from pulmonary embolism. In any hypotensive patient, induction medications can result in hemodynamic decompensation and/or cardiac arrest. Further, a shock index (heart rate/systolic blood pressure) greater than 1.0 is an independent predictor of peri-intubation cardiac arrest.[ix] Ketamine and etomidate appear to be the most hemodynamically favorable agents for induction.[x] No randomized studies have demonstrated superiority of one of these agents over the other.
Improving Cardiac Output
Improving systemic blood pressure in acute PE helps improve coronary blood flow to the RV and prevents right ventricular ischemia. Numerous factors have deleterious effects on systemic blood pressure and cardiac output in the setting of massive PE. Increases in intrathoracic pressure from positive pressure ventilation negatively affect cardiac output. High intrathoracic pressures impair venous return and reduce preload. It is unlikely that intravenous fluid loading will have a significant positive effect on preload in a patient who has hemodynamic compromise from acute PE. In severe RV dysfunction, there is a chance that intravenous fluid loading may actually hasten RV failure. The increase in venous return may worsen RV dilation, RV ischemia and septal shift, and further decrease LV end-diastolic volume.[xi],[xii]
Vasoactive medications are often required for the treatment of shock due to acute PE. The optimal vasopressor is unknown, but norepinephrine is often the initial vasopressor of choice.[xiii] Norepinephrine acts on both alpha-1 and beta-1 receptors, resulting in peripheral vasoconstriction and a modest increase in cardiac output. Norepinephrine also improves blood flow to cardiac myocytes, resulting in further inotropy.[xiv] Phenylephrine, a pure alpha agonist, also increases systemic blood pressure but likely increases PVR more than norepinephrine, decreasing its usefulness in pulmonary embolism.[xv] If norepinephrine fails to improve shock, dobutamine is often added. Dobutamine is not a vasopressor, but a pure inotrope that activates the beta-1 adrenergic receptor. This results in increases in inotropy and chronotropy. It has minimal alpha-1 and beta-2 effects, resulting in a reflex vasodilation. This vasodilation can be mitigated by the concomitant use of norepinephrine. Although milrinone is another potent inotropic agent, its profound vasodilatory properties and prolonged half-life can limit its utility in the setting of profound hypotension.[xvi]
Initiating vasoactive medications early in the course of PE-induced hemodynamic collapse is of paramount importance. Waiting to obtain central intravenous access may result in further deterioration. Newer studies suggest that initiating vasopressors through a proximal peripheral IV for less than four hours duration is unlikely to result in tissue injury and will reduce the time it takes to achieve hemodynamic stability.[xvii]
In severe shock, vasoactive medications may be unable to restore systemic perfusion. In these situations, consideration should be given for initiation of venoarterial (VA) ECMO. This may be a reasonable treatment approach in patients with refractory shock who have either failed or have contraindications to systemic thrombolysis. Since over 90% of patients respond favorably to thrombolysis with 36 hours,[xviii] VA-ECMO may serve as a bridge to recovery or further intervention.
Cardiac Arrest and Suspected Pulmonary Embolism
When circulatory collapse becomes too severe, cardiac arrest may occur. It is estimated that a substantial number of sudden cardiac arrests are caused by pulmonary embolism.[xix] In the setting of cardiac arrest or severe hypotension, immediate CT angiography may not be feasible. In this circumstance, the most important diagnostic tool is bedside echocardiography. The finding of RV dysfunction in this clinical scenario is enough to prompt primary reperfusion. Emergency physicians can reliably perform and interpret focused, goal-directed echocardiography.[xx] Part of this assessment is a gross visual estimate of RV size and function. Occasionally, visualization of free-floating thrombi in the right atrium or ventricle may solidify the diagnosis of PE. In the absence of findings of right ventricular dysfunction in cardiac arrest, empiric thrombolysis should not routinely be administered.
Case Resolution
Case 1
This patient presents with acute chest pain/dyspnea and is found to have a pulmonary embolism. Because of persistent hypotension, she is classified as a massive PE. The goals of her resuscitation are improvement of hypoxia, acute clot reduction, and improvement in systemic hypotension. These can be performed rapidly and simultaneously with the administration of supplemental oxygen (with or without inhaled nitric oxide), prompt initiation of heparin anticoagulation and systemic thrombolysis, and initiation of vasoactive medications (norepinephrine +/- dobutamine). If the patient has refractory hypotension, further consideration should be given to catheter-directed thrombolysis or surgical embolectomy.
Case 2
This patient presents with acute onset dyspnea. Initially, his blood pressure is normal but he soon develops circulatory collapse. The most important diagnostic modality is bedside echocardiography. The concept of Rapid Ultrasound for Shock and Hypotension (RUSH) was developed to aid in the diagnosis of undifferentiated hypotension.[xxi] Part of the RUSH exam involves a gross estimate of RV size. In the setting of severe hypotension, the finding of RV dysfunction is enough to prompt rapid reperfusion.
References/Further Reading
[i] Stein PD, Beemath A, Matta F, Weg JG, Yusen RD, Hales CA, Hull RD, Leeper KV Jr, Sostman HD. Clinical characteristics of patients with acute pulmonary embolism: data from PIOPED II. Am J Med. 2007;120(10):871.
[ii] Jaff et al. AHA Scientific Statement: Management of Massive and Submassive Pulmonary Embolism, Iliofemoral Deep Vein Thrombosis, and Chronic Thromboembolic Pulmonary Hypertension. Circulation, 2011; 123: 1788-1830.
[iii] Dalen JE, Alpert JS. Natural history of pulmonary embolism. Prog Cardiovasc Dis. 1975;17(4):259.
[iv] Kearon C, Akl EA, Comerota AJ, Prandoni P, Bounameaux H, Goldhaber SZ, Nelson ME, Wells PS, Gould MK, Dentali F, Crowther M, Kahn SR, American College of Chest Physicians. Antithrombotic therapy for VTE disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141.
[v] Piazza G, Hohlfelder B, Jaff MR, et al. A Prospective, Single-Arm, Multicenter Trial of Ultrasound-Facilitated, Catheter-Directed, Low-Dose Fibrinolysis for Acute Massive and Submassive Pulmonary Embolism. The SEATTLE II Study. J Am Coll Cardiol Intv. 2015;8:1382.
[vi] Stein PD, Alnas M, Beemath A, Patel NR. Outcome of pulmonary embolectomy. Am J Cardiol. 2007 Feb;99(3):421-3. Epub 2006 Dec 15.
[vii] Bhat T et al. Inhaled nitric oxide in acute pulmonary embolism: a systematic review. Rev Cardiovascular Med. 2015;16(1):1-8.
[viii] Burrowes KS, Clark AR, Tawhai MH. Blood flow redistribution and ventilation-perfusion mismatch during embolic pulmonary arterial occlusion. Pulm Circ 2011;1(3):365–376.
[ix] Heffner AC, Swords DS, Neale MN, Jones AE. Incidence and factors associated with cardiac arrest complicating emergency airway management. Resuscitation. 2013 Nov;84(11):1500-4. Epub 2013 Aug 1.
[x] Morris C, Perris A, Klein J, Mahoney P. Anaesthesia in haemodynamically compromised patients: does ketamine represent the best choice of induction agent? Anaesthesia. 2009 May;64(5):532-9.
[xi] Belenkie I, Dani R, Smith ER, Tyberg JV. Ventricular interaction during experimental acute pulmonary embolism. Circulation. 1988;78:761–768.
[xii] Belenkie I, Dani R, Smith ER, Tyberg JV. Effects of volume loading during experimental acute pulmonary embolism. Circulation. 1989;80:178–188.
[xiii] Ghignone M, Girling L, Prewitt RM. Volume expansion versus norepinephrine in treatment of a low cardiac output complicating an acute increase in right ventricular afterload in dogs. Anesthesiology. 1984;60(2):132.
[xiv] Practice parameters for hemodynamic support of sepsis in adult patients in sepsis. Task Force of the American College of Critical Care Medicine, Society of Critical Care Medicine. Crit Care Med. 1999;27(3):639.
[xv] Rich, S, Gubin S, Hart K. The effects of phenylephrine on right ventricular performance in patients with pulmonary hypertension. CHEST. 1990 Nov;98(5):1102-6.
[xvi] Lollgen H, Drexler H. Use of inotropes in the critical care setting. Crit Care Med. 1990;18(1 Pt 2):S56.
[xvii] Loubani OM, Green RS. A systematic review of extravasation and local tissue injury from administration of vasopressors through peripheral intravenous catheters and central venous catheters. J Crit Care. 2015 Jun;30(3):653.
[xviii] Meneveau N, Seronde MF, Blonde MC, Legalery P, Didier-Petit K, Briand F,
Caulfield F, Schiele F, Bernard Y, Bassand JP. Management of unsuccessful thrombolysis in acute massive pulmonary embolism. Chest 2006;129(4):1043–1050.
[xix] Corness KA, DeRook FA, Russell ML, Tognazzi-Evans TA, Beach KW. The incidence of pulmonary embolism in unexplained sudden cardiac arrest with pulseless electrical activity. Am J Med. 2000 Oct 1;109(5):351-6.
[xx] Jones AE, Tayal VS, Kline JA. Focused training of emergency medicine residents in goal-directed echocardiography: a prospective study. Acad Emerg Med. 2003 Oct;10(10):1054-8.
[xxi] S. D. Weingart, D. Duque, and B. Nelson, Rapid Ultrasound for Shock and Hypotension (RUSH-HIMAPP), 2009, http://emedhome.com/.





4 thoughts on “Pulmonary Embolism: Management of the Unstable Patient”
Pingback: LITFL Review 213 | LITFL: Life in the Fast Lane Medical Blog
Pingback: Länkar v1 | Internmedicin
Pingback: Pulmonary Embolism | Emergency Medicine Cases : Emergency Medicine Cases
Pingback: Ciężka Zatorowość Płucna — Twój Dyżur — Kurs, Przypadki, Dyskusje