Author: Erica Simon, DO, MHA (EM Resident Physician, SAUSHEC) // Edited by: Manpreet Singh, MD (@MPrizzleER – Clinical Instructor & Ultrasound/Med-Ed Fellow / Harbor-UCLA Medical Center) and Alex Koyfman, MD (@EMHighAK, EM Attending Physician, UT Southwestern Medical Center / Parkland Memorial Hospital)
 A 4-year-old African American female presents to your ED in acute respiratory distress. First responders quickly relay their account of a young female, struggling for air, with bilateral rales and an SpO2 of 82% on RA. The remainder of the triage VS are reported as HR 144, RR 47, BP 85/62, T 102.4 oral.
A 4-year-old African American female presents to your ED in acute respiratory distress. First responders quickly relay their account of a young female, struggling for air, with bilateral rales and an SpO2 of 82% on RA. The remainder of the triage VS are reported as HR 144, RR 47, BP 85/62, T 102.4 oral.
Supplemental oxygen therapy (non-rebreather), initiated en route, has improved the patient’s SpO2 to 91%. Your young patient is leaning forward in her bed gasping. She has marked supraclavicular and intercostal retractions, and you note the previously mentioned rales. The patient’s mother, having joined EMS personnel on the way to your facility, reports a PMHx of sickle cell anemia (SCA).
What is your plan of action? CXR? Antibiotics? Intubate? Hopefully by the end of this quick review you’ll have a few pearls to reference.
The Pathophysiology of SCD
Let’s start at the beginning (i.e. – knowledge you were happy to leave behind s/p STEP2):
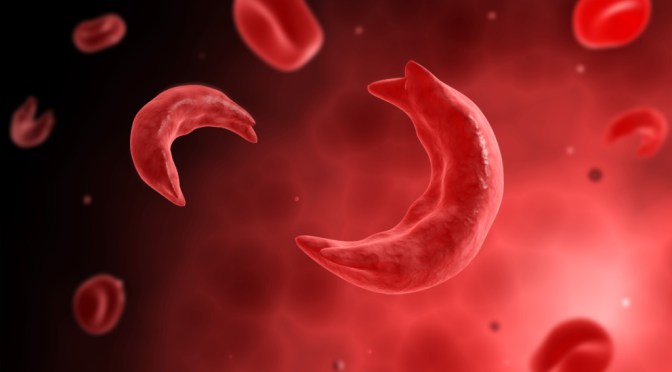
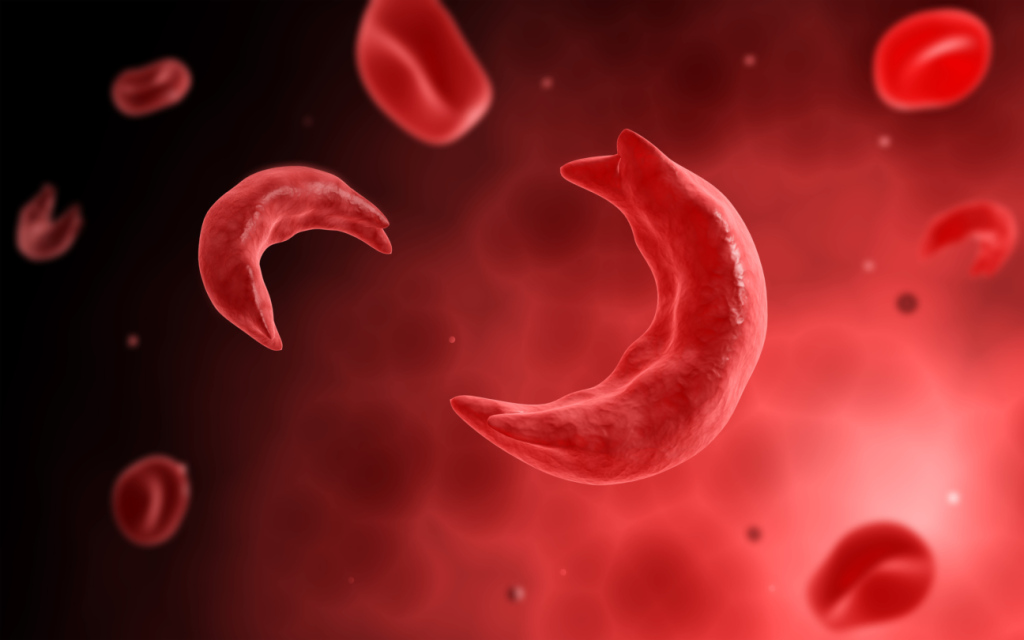
The sickle cell mutation, a sixth codon substitution of the B-globin chain (exchanging valine for glutamic acid), alters the structural conformation of the B-globin molecule under deoxygenated conditions, resulting in a sickled appearance.1,2
The mutation is inherited in an autosomal recessive fashion. Homozygotes are said to exhibit sickle cell anemia (SCA or HBSS) and heterozygotes, sickle cell trait (SCT). Assuming that they have not inherited a second abnormal hemoglobin (Hb) chain, individuals with SCT are commonly asymptomatic, while those with SCA are predisposed to severe infections, capillary obstruction/painful vaso-occlusive crisis and multi-system organ damage.2
The Epidemiology and Presentation of SCD
SCD affects nearly 100,000 individuals in the US, and approximately 2 million Americans carry the sickle cell trait.2,3 SCA is prevalent in persons of African, Mediterranean, Indian, and Middle Eastern descent.2
As SCD is a component of the American newborn screen, it’s unlikely that you’ll make the diagnosis in the ED. Most commonly, individuals with SCA will present for evaluation after the fourth month of life (decline in levels of fetal Hb), 2/2 to sequelae of chronic anemia (jaundice, fatigue, FTT) vs. acute complications of the disease (vaso-occlusive crisis, splenic sequestrations, stroke, etc).2
Managing Acute Complications of SCA
Vaso-occlusive Phenomena
Stroke
Ischemic stroke and intracranial hemorrhage are major complications of SCD. As you might expect, clinical presentation varies according to the anatomic location of the infarct/hemorrhage. Small infarcts are relatively common and involve the basal ganglia and deep white matter within the anterior circulation.4 Risk factors for stroke in patients with SCD include: high flow velocity on transcranial Doppler (see below), low Hb, high WBC count, hypertension, hx of silent infarcts, and hx of acute chest syndrome.4
Transcranial Doppler: strongly recommended as an annual screening exam in patients ages 2-16 years to determine candidacy for long-term transfusion therapy for stroke prevention (abnormal velocity ≥ 200cm/s).3
Evaluation: Neuroimaging is key. Keep this in your back pocket: a CT scan will miss early signs of ischemic infarction and is insufficient for ruling out stroke in a child. MRI with diffusion-weighted imaging and MRA of the head and neck, should be performed in all pediatric patients with suspected acute ischemic stroke.5
Treatment: Adult patients with ischemic strokes should undergo evaluation for tPA candidacy vs. intra-arterial thrombolysis. Pediatric patients with SCD should receive IVFs and exchange transfusion to achieve a hemoglobin S level of <30%.5 If an exchange transfusion can’t be arranged, a simple transfusion should be performed.5
Keep in mind:
- Target a MAX Hb of 13g/dL s/p transfusion – peds with SCD may be at risk for recurrent ischemia secondary to increased blood viscosity.5
- Hemorrhagic transformation occurs in 30% of children with arterial ischemic stroke, and is frequently asymptomatic.6
Addressing HTN: In children, hypertension (i.e., blood pressure >95th percentile for age) within the first 72 hours after ischemic stroke is associated with an increased risk of death.7 A blood pressure goal of the 50th-95th percentile for age and height, with permissive HTN up to 20% above the 95th percentile should be targeted.5 Experts recommend the use of labetolol or an ACEI to lower BP by 25% over 24 hours.5 (Caution here – evaluate renal function prior to initiating an ACEI, we’ll discuss the reason shortly).
PEARLS: Do not forget to investigate alternative causes of stroke: infection, cardiac embolism, cavernous venous sinus thrombosis, etc. Seizures are COMMON s/p pediatric neurological injury.8 Patients with persistent lethargy should be evaluated with EEG for subclinical seizure activity.5
Acute Chest Syndrome (ACS)
ACS is a leading cause of morbidity and mortality among patients with SCD.9 Patients may present with fever, cough, chest pain, hemoptysis, or dyspnea. While the pathogenesis of ACS has yet to be determined, infection frequently represents the underlying etiology in the pediatric population.10 The evaluation of ACS should include a CXR (to evaluate for the presence of a new infiltrate), SpO2 monitoring, and a CBC (to assess for anemia).
Evidence-based guidelines, and expert panels recommend the following in the management and treatment of ACS:
- All patients with ACS should be hospitalized for pain control and SpO2 monitoring. (Consensus-Panel Expertise)3,11
- Patients with SCD who have ACS should receive antibiotics (parenteral cephalosporin therapy and oral macrolide therapy). (Strong Recommendation, Low-Quality Evidence)3,10
- Patients should receive supplemental O2 to maintain SpO2 > 95%. (Strong Recommendation, Low-Quality Evidence)3,11
- SCA patients with ACS should receive a blood transfusion to improve O2 carrying capacity if Hb is >1g/dL below baseline. If baseline is ≥9g/dL, transfusion may not be required. (Weak Recommendation, Low-Quality Evidence)3,11
- In all persons with SCD, urgent exchange transfusion, in consultation with a specialist, should be performed when there is rapid progression of ACS: SpO2 <90 % despite supplemental oxygen, increasing respiratory distress, progressive pulmonary infiltrates, and/or decline in hemoglobin concentration despite simple transfusion. (Strong Recommendation, Low-Quality Evidence)3,11
Beware: ACS may rapidly progress to ARDS 2/2 pulmonary sequestration/infarct. Your disposition = ICU. The severity of illness and the mortality rate are greater in adults than children (4.3% vs. 1.8%, respectively).10 Long-term complications of ACS include pulmonary fibrosis, pulm HTN, and cor pulmonale.
Note: Please review references 3 and 11 for an in-depth discussion of evidence strength and expert panel qualifications.
Acute Pain Crisis
Vaso-occlusive pain crises may manifest in a number of systems: pulmonary system (CP/SOB), CNS (HA), skeletal system (arthralgias/dactylitis), GI system (abdominal pain). In the setting of these crises, patients commonly present with fever and leukocytosis.10 The key is to rule out SCD manifestations that require emergent intervention and to treat pain EARLY. For example:
CP/SOB? => ACS vs. ARDS vs. Cor Pulmonale vs. high-output heart failure 2/2 anemia vs. PNA vs. pain crisis
HA => stroke vs. meningitis 2/2 an encapsulated organism (discussed below) vs. cerebral venous sinus thrombosis vs. ocular pathology vs. pain crisis
Arthralgias => septic arthritis vs. acute trauma vs. avascular necrosis 2/2 sickling vs. pain crisis
Abdominal pain => splenic sequestration vs. acute intrahepatic sequestration vs. pain crisis 2/2 occlusion of mesenteric vasculature… or is it really flank pain => renal infarct 2/2 vaso-occlusion (discussed below)
Keep your differential broad as you approach a sickler with the CC of “pain crisis,” and always question the patient regarding “normal patterns” of previous crises. Any atypical pain pattern requires evaluation.3
Evidence-based guidelines and expert panels recommend the following in the management and treatment of pain crisis:
- Initiate analgesia within 30 minutes of triage. (Consensus – Panel Expertise) 3,11
- Employ individualized prescribing and pain monitoring protocols. (Consensus – Panel Expertise) 3,11
- If no contraindications, give NSAIDs as adjuvant pain therapy. (Moderate Recommendation, Low-Quality Evidence) 3,11
- Avoid meperidine (normeperidine, the active metabolite of meperidine, is excreted by the kidneys, and is associated with an increased incidence of seizures in the setting of renal dysfunction – common in occlusive crisis)3,10,11
Sequelae of Hemoglobinopathy – The Abdomen
RUQ Pain
RUQ pathology is common in SCD. The challenge for the EM physician is to determine if the etiology is symptomatic cholelithiasis vs. cholecystitis vs. acute intrahepatic cholestasis (AIC) vs. acute sickle hepatic crisis vs. acute hepatic sequestration (AHS). CBC, LFTs, coags, and imaging (CT vs. US) are a must.1
A few words on each condition:
- Pigmented gallstones occur in 30-70% of patients.2
- AIC is a result of sickled RBCs occluding hepatic sinusoids, causing vascular stasis and local hypoxia. As Kupffer cells (hepatic macrophages) phagocytose sickled erythrocytes, hepatic real estate becomes sparse and canniliculi occlude with bile.12 Patient presentation ranges from isolated hyperbilirubinemia with preserved hepatic function (PT/aPTT WNL) to RUQ pain, transaminitis, and extreme elevations of bilirubin and alk phos. In the latter case, renal failure, thrombocytopenia, and severely prolonged coagulation times often develop.13 If you suspect this – consult early, exchange transfusion may be indicated.13
- Acute sickle hepatic crisis affects 10% of patients admitted for painful crisis.13 The crisis mimics acute cholecystitis with RUQ pain, fever, leukocytosis, and variable increases in serum transaminases and bilirubin levels, however, unlike cholecystitis, hepatomegaly occurs.13 Treatment is supportive with pain control and consultation for possible transfusion.1,13
- AHS occurs 2/2 obstruction of sinusoidal flow by masses of sickled erythrocytes and can be a complication of acute sickle hepatic crisis.12,13 In addition to RUQ pain, fever, jaundice, and hepatomegaly, labs reveal an acute drop in H&H with reticulocytosis.13 Similar to splenic sequestration – consult. This too can be an indication for simple or exchange transfusion.1,14
PEARL: The laboratory studies of a patient with SCA cannot be interpreted in a vacuum. Obtaining a PMHx regarding transfusions, baseline hepatic function/diagnosed (CT) micro-infarcts is invaluable in determining chronic vs. acute hepatic pathology. (Chronic liver disease might be 2/2 hemosiderosis and possibly due to the SCD itself through clinically silent microvascular occlusions.)13
Splenic Sequestration
Pooling of RBCs in splenic sinusoids may result in splenic sequestration, or an acute drop in hemoglobin levels (> 2 g/dL) associated with splenomegaly, reticulocytosis, and signs of intravascular volume depletion.10 Sequestration can RAPIDLY progress to shock and death. Drops in hemoglobin levels > 4 g/dL are associated with 35% mortality in the pediatric population.9,10
Splenic sequestration typically occurs in kids 10-27 months of age, but may be seen as early as 2 months of age.9,10 Emergency management is aimed at restoring circulating blood volume: IVF resuscitation and simple/exchange blood transfusions in consultation with specialists. 9,10,11 As splenic sequestration has a high rate of recurrence, all cases should be managed in conjunction with a hematologist as patient may be considered for splenectomy.1,10 After 3 to 5 years of age, the risk of splenic sequestration decreases dramatically owing to splenic auto-infarction.
Sequelae of Hemoglobinopathy – The GU System
Sickle Cell Nephropathy/Infarcts
The kidney is one of the most commonly affected organs in SCD.15 Ischemic damage caused by RBC sickling in the vasa recta predisposes patients to a number of glomerulopathies, resulting in renal dysfunction.15 Infarct of the renal medulla secondary to vaso-occlusion may cause flank pain and CVA tenderness. Alternatively, papillary necrosis may present with gross or microscopic hematuria.2 Imaging is required to make a diagnosis (CT with IV contrast vs. CT IVP), and both renal medulla infarction and papillary necrosis are treated with IV fluids and admission.2,16 Ultimately 30% of adult patients with SCD develop chronic kidney disease with approximately 12% of patients going on to develop ESRD.16,17
Priapism
SCD accounts for >60% of ischemic (low-flow) priapism cases in children and >25% of cases in adults.16 Treatment of ischemic priapism includes needle aspiration of blood from the corpora cavernosa, followed by intercavernosal injection of 1ml aliquots (up to 3 ml) of 100-500ug/ml phenylephrine.2,16 Measures to treat sickle cell disease (hydration, oxygen, exchange transfusions) may be utilized, but should not delay aspiration and phenylephrine injections.16
Infection
Fourteen percent of SCD patients are functionally asplenic (2/2 autoinfarction) by 6 months of age; this number reaches 94% by the 5 years of age.2 (Note: a number of SCA patients may also be s/p splenectomy 2/2 recurrent sequestration crisis). S. pneumoniae, H. influenzae type b, non-typhi Salmonella species, Mycoplasma, C. pneumoniae and Yersinia enterocolitica are important pathogens in sickle cell patients.14 In order to prevent serious infection due to encapsulated organisms, preventative measures are recommended for patients with SCD:
- Prophylactic PCN V for patients age 2 months – 5 years to prevent SBI.1
- Pneumococcal vaccine at 2 months of age to reduce the risk of pneumococcal infection.1
- Influenza vaccination at 6 months and annually1
- Meningococcal vaccination for children with splenic dysfunction at 2 years of age. 1
This is an important history to elucidate as a fever in a SCD patient is a true emergency – common etiologies include bacteremia, osteomyelitis, or infection causing ACS.17 As mentioned above, pain crisis can also present with fever, always be on the lookout for a source.
Treatment of Presumed Infection in SCD (Adapted)17

Note: Targeted antibiotic therapy for osteomyelitis should cover salmonella and s. aureus.
Aplastic Crisis
Aplastic crisis in SCA is commonly secondary to Parvovirus B19 infection. Patients may present with pallor and tachycardia due to a transient failure of erythropoiesis.2 Emergency department care is supportive and depends upon the degree of anemia and cardiovascular compromise.10 If the reticulocyte count is less than 1% to 2% with no signs of spontaneous recovery, simple transfusions are administered to raise the hemoglobin to approximately 10 g/dL and the hematocrit to about 30%.10,18 SCA patients with aplastic crisis should be admitted to the ICU setting with droplet precautions.
A Quick Word on Transfusion
The Jenkins article offers a great summary of the ins and outs of transfusion indications10:

Transfusion Indications10
References / Further Reading
- Lanzkron S, Domm J, Sweet K. Sickle cell disease. First Consult. Elsevier BV, 2010.
- Williams-Johnson J, Williams E. Chapter 231: Sickle cell disease and other hereditary hemolytic anemias. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide 7e. Chapel Hill, NC, McGraw-Hill Holdings, LLC, 2011.
- Yawn B, Buchanan G, Afenyi-Annan A, Ballas S, Hassell K, et al. Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members. JAMA. 2014; 312(10): 1033-1048.
- Roach S, Golomb M, Adams R, Biller J, Daniels S, et al. Management of stroke in infants and children. American heart association: scientific statement. Stroke. 2008; 39: 2644-2691.
- Elbers J, Wainwright M, Amlie-Lefond C. The pediatric stroke code: early management of the child with stroke. J Pediatr. 2015; 167(1): 19-24e4.
- Beslow LA, Smith SE, Vossough A, Licht DJ, Kasner SE, Favilla CG, et al. Hemorrhagic transformation of childhood arterial ischemic stroke. Stroke. 2011;42:941-946.
- Brush LN, Monagle PT, Mackay MT, Gordon AL. Hypertension at time of diagnosis and long-term outcome after childhood ischemic stroke. Neurology. 2013;80:1225-1230.
- Abend NS, Gutierrez-Colina AM, Topjian AA, Zhao H, Guo R, Donnelly M, et al. Nonconvulsive seizures are common in critically ill children. Neurology. 2011;76:1071-1077.
- Eskenazi AE, Bertstein MC, Gordon JB. Hematologic disorders in the pediatric intensive care unit. In: MC Rogers, ed. Textbook of Pediatric Intensive Care. 3rd ed. Baltimore, Md: Lippincott Williams & Wilkins; 1997: 1395–1431.
- Jenkins, T. Sickle cell anemia in the pediatric intensive care unit: novel approaches for managing life-threatening complications. AACN Clinical Issues. 2002; 13(2): 154-168.
- Afenyi-Annan A, Ballas S, Hassell K, James A, Jordan L, et al. Managing acute complications of sickle cell disease: evidence-based management of sickle cell disease. National Heart, Lung, and Blood Institute. 2014; 31-54.
- Norris W. Acute hepatic sequestration in sickle cell disease. J Natl Med Assoc. 2004; 96(9):1235-1239.
- Ebert E, Nagar M, Hagspiel K. Gastrointestinal and hepatic complications of sickle cell disease. Clinical Gastroenterology and Hepatology: American Gastroenterology Association. 2010; 8(6): 483-489.
- Johnson C, Omata M, Tong M, Simmons, J, Weiner J, et al. Liver involvement in sickle cell disease. Medicine (Baltimore) 1985;64:349-356.
- Gebreselassie S, Simmons M, Montague D. Genitourinary manifestations of sickle cell disease. CCJM 2015; 82(10): 679-683.
- Bruno D, Wigfall D, Zimmerman S, et al: Genitourinary complications of sickle cell disease. J Urol 166: 803, 2001.
- Sobota A, Sabharwal V, Fonebi G, and Steinberg M. How we prevent and manage infection in sickle cell disease. Br J Haematol. 2015; 170(6): 757-767.
- Rogers ZR, Nickerson BG. Life-threatening complications in sickle cell disease. In: DL Levin, FC Morriss, eds. Essentials of Pediatric Intensive Care. 2nd ed. Vol. II. New York: Churchill Livingstone, Quality Medical Publishing; 1997:483–487.
- http://www.ncbi.nlm.nih.gov/pubmed/25060254