Thrombotic Thrombocytopenic Purpura: Pearls and Pitfalls
By: Brit Long, MD
EM Resident Physician at SAUSHEC; USAF
Edited by: Alex Koyfman, MD (@EMHighAK, EM Attending Physician, UTSW Medical Center / Parkland Memorial Hospital) and Stephen Alerhand, MD (@SAlerhand)
A 72 year-old female presents with altered mental status and decreased urine output from her nursing home. Initial vital signs reveal a temperature of 100.5, but are otherwise normal. Exam reveals an elderly cachectic female who is disoriented to place, situation, and date. She has dry mucous membranes, but otherwise the rest of her exam (including neuro) is normal. Lab results shows a creatinine of 2.5, elevated from her 0.8 baseline, along with a hemoglobin of 7.2 and platelets of 24,000.
Altered mental status in an elderly adult has a wide differential. However, these laboratory findings of anemia, thrombocytopenia, and acute renal injury suggest thrombotic thrombocytopenic purpura (TTP) as the primary etiology.
Introduction
TTP is a rare disease but without treatment, mortality reaches close to 100%. With treatment, mortality drops to 10-20%.1-3 The annual incidence worldwide is 3.7 per year per million people, with a female predominance.1-4
Pathophysiology
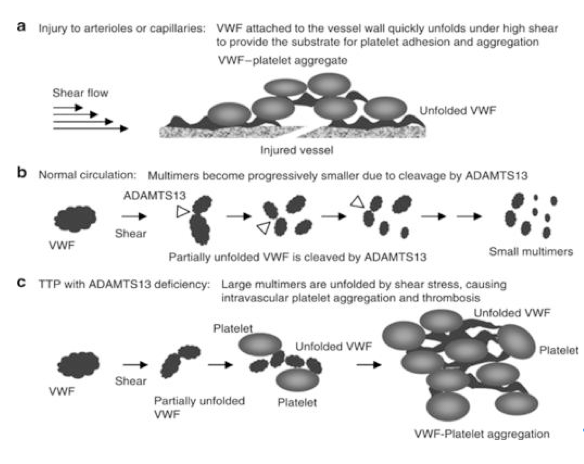
Symptoms are due to endothelial injury with platelet rich thrombi causing microangiopathic hemolytic anemia (MAHA) and thrombocytopenia. Von Willebrand factor (vWF) is made in endothelial cells and assembled in large multimers. These large multimers are cut by ADAMTS13 into smaller units. If ADAMTS13 protease does not function correctly, multimers of vWF collect and accumulate (Of note, ADAMTS13 deficiency can be either inherited or acquired through an autoimmune mechanism.). Platelets attach and promote aggregation, which form clots. These multimers also cleave RBCs intravascularly, resulting in the classic MAHA. Symptoms and signs result from these platelet plugs and MAHA. Normal ADAMTS13 activity is greater than 50%. If the activity is normal, other pathologies are likely present including: autoantibodies to ADAMTS13, complement activation, ADAMTS13 inhibitor antibody, or plasminogen activator malfunction. Inciting triggers include infection (HIV), malignancy, pregnancy, medication, organ transplantation, chemotherapy, pancreatitis, autoimmune disease, or most commonly, an idiopathic cause.
Presentation
The classic presentation consists of a pentad – a neurologic finding, low platelets with purpura, microangiopathic hemolytic anemia (MAHA), fever, and acute renal failure/injury.1-6 However, this pentad is present 1/3 of the time!7 The classic pentad was more common in the past when patients would present in fulminant TTP. However, many patients today present before this state. The presentation of microangiopathic anemia, thrombocytopenia, and some neurologic deficit is present in ¾ of patients.7
- Neurologic manifestations range from headache to coma, with seizures included, and these symptoms usually fluctuate. Focal deficits are less prevalent. Anywhere from 25 to 60% of patients experience a neurologic manifestation. These findings are usually transient and subtle, so you must ask the patient about prior symptoms! The transient nature is due to unstable aggregation of platelets in the central nervous system.
- Thrombocytopenia is a major diagnostic criterion for TTP. Studies demonstrate a mean platelet count of 25,000. However, the platelets may reach 5,000. Interestingly, in patients with severe renal disease the thrombocytopenia is often not as severe!
- MAHA is a second important criterion, and this is defined as non-immune hemolysis with schistocytes resulting from RBC fragmentation. This causes elevated LDH, low haptoglobin, high indirect bilirubin. LDH is often severely elevated, and this lab value can be used to evaluate response to treatment!
- Fever is uncommon in patients with TTP if they present early in the disease course. Fever is normally present ¼ of the time. High fever and chills suggest sepsis, so look for a source of infection in these patients!
- Renal disease is due to renal thrombotic microangiopathy, with urinalysis showing mild proteinuria with casts, and at times hematuria. This is usually due to endothelial injury from complement activation. Acute renal insufficiency may present with anuria and often requires dialysis.
- Other symptoms include abdominal complaints such as pain, nausea, vomiting, or diarrhea. The patient will often have splenomegaly and jaundice. Cardiac involvement includes MI, arrhythmia, shock, and heart failure. In one study with TTP, 10% of patients suffered from acute heart failure. Elevated LDH is associated with acute MI in patients, specifically an LDH above 100.2-9
Risk Factors
Obesity, African American race, female, patients age 30 to 50 years, HIV/AIDS, rheumatologic/autoimmune disease history, and clopidogrel (Plavix) all increase risk of developing TTP.7
Diagnosis
In any patient with MAHA and thrombocytopenia, consider TTP and consult hematology. This is the most important aspect of this disease! Lab findings will include low hemoglobin, low platelets, elevated LDH, potentially a decrease in renal function and electrolyte abnormalities, and elevated bilirubin (indirect). A peripheral smear is vital for diagnosis, as 100% of patients will have schistocytes present during the course of the disease. Urine may show proteinuria, hematuria, and/or casts, which are dependent on the state of renal injury. Coagulation tests will be normal or minimally elevated, which is a differentiating factor between TTP and DIC (markedly elevated coagulation tests). Your hematologist will want ADAMTS13 activity. A finding of activity < 10% is supportive of TTP diagnosis, but this is not definitive.Remember, TTP is a clinical diagnosis. Normal activity is above 50%. If the activity level is normal, Evan’s syndrome may be present, which is MAHA with thrombocytopenia and no other abnormalities/symptoms.2-10
Differential
Bloody diarrhea caused by Shiga-toxin producing bacteria is more common in the pediatric population due to hemolytic uremic syndrome (HUS), but bloody diarrhea can be due to mesenteric ischemia in TTP. In pregnant patients, HELLP syndrome will have greater LFT increase, whereas in TTP the LFTs will be normal or only mildly elevated. DIC will have abnormal coagulation studies. Patients with systemic vasculitis may present similarly and will ultimately need tissue biopsy for confirmation. A disseminated malignancy can present in a similar manner.7
Treatment
The first aspect of treatment is avoid transfusing platelets if possible! Platelet transfusion is associated with increased morbidity in TTP, specifically higher rates of arterial thrombosis (but not venous thrombosis) as this provides further platelets to form larger clots. A large systematic literature review suggested that harm from platelet transfusion was uncertain.12 If needed for procedure or severe bleeding, platelet transfusion should occur (such as need for lumbar puncture in the setting of low platelets).2-12
The goal is hematology consultation, as ultimately these patients need plasma exchange/plasmapheresis. This treatment works to filter circulating antibodies against ADAMTS13 and the large, platelet-coated ADAMTS13 multimers.13 The treatment usually requires a central line or two large IVs if central line access is unavailable. RBCs are filtered from plasma and returned with donated FFP and albumin. The patient’s plasma with antibodies and ADAMTS13 multimers is removed and discarded. This is completed with some form of semipermeable based filtration system. Response to treatment can be followed by improving symptoms and labs (such as improved LDH, bilirubin, and platelet levels). This treatment is done daily and then tapered with resolving laboratory and clinical improvement to several times per week.2-4,7,13
If plasma exchange is not immediately available, transfusion of FFP can be helpful, though studies have shown this is not definitive treatment. Roughly 10 to 20% of patients will not respond properly to exchange. Steroids are an adjunct treatment and do not replace plasmapheresis, as relapse is certain in those treated with steroids and not exchange, and side effect rate is high. IVIG is also not first line, but may be used in those who fail plasmapheresis. Immunomodulators (vincristine, cyclophosphamide, rituximab, etc.) are only supported in treatment failures by case reports, and last line is splenectomy.2-4,7,13
Outcome
Unfortunately relapse is seen in up to 50% of cases, and it is defined as new onset after 30 days of remission. It is most commonly seen in the first two years following remission, and those patients with ADAMTS13 activity less than 10% are at risk.2,3,7,14
Summary
-TTP is a clinical diagnosis: suspect TTP in a sick-appearing patient with MAHA and thrombocytopenia.
-TTP is due to deficiency/inactivity of ADAMTS13.
-The classic pentad is rare, but you can depend on MAHA and low platelets for diagnosis.
–Schistocytes are present in 100% of cases at some point in the disease course.
-Treatment is plasma exchange. FFP can assist if exchange is delayed.
–Avoid platelet transfusion if possible.
References/Further Reading
- Moake J.L.: Thrombotic microangiopathies. N Engl J Med 2002; 347: pp. 589-600.
- Comparison of plasma exchange with plasma infusion in the treatment of thrombotic thrombocytopenic purpura. N Engl J Med 1991; 325: pp. 393-397.
- Allford S.L., Hunt B.J., Rose P., Hemostasis and Thrombosis Task Force , British Committee for Standards in Hematology , et al: Guidelines on the diagnosis and management of the thrombotic microangiopathic hemolytic anemias. Br J Hematol 2003; 120: pp. 556-573.
- Scully M., Hunt B.J., Benjamin S., et al: Guidelines on the diagnosis and management of thrombotic thrombocytopenic purpura and other thrombotic microangiopathies. Br J Hematol 2012; 158: pp. 323-335.
- Interventions for hemolytic uremic syndrome and thrombotic thrombocytopenic purpura. Cochrane Database Syst Rev
- Coppo P., and Veyradier A.: Current management and therapeutical perspectives in thrombotic thrombocytopenic purpura. Presse Med 2012; 41: pp. 163-176.
- Kessler C.S., Khan B.A., and Lai-Miller K.: Thrombotic thrombocytopenic purpura: a hematological emergency. J Emerg Med 2012; 43: pp. 538-544.
- George J.N.: How I treat patients with thrombotic thrombocytopenic purpura: 2010. Blood 2010; 116: pp. 4060-4069.
- Hughes C., McEwan J.R., Longair I., et al: Cardiac involvement in acute thrombotic thrombocytopenic purpura: association with troponin T and IgG antibodies to ADAMTS 13. J Thromb Haemost 2009; 7: pp. 529-53.
- Tsai H.-M., and Lian E.C.: Antibodies to von-Willebrand factor-cleaving protease in acute thrombotic thrombocytopenic purpura. N Engl J Med 1998; 339: pp. 1585-1594.
- Furlan M., Robles R., Galbusera M., et al: Von Willebrand factor–cleaving protease in thrombotic thrombocytopenic purpura and the hemolytic–uremic syndrome. N Engl J Med 1998; 339: pp. 1578-1584.
- Duffy S.M., and Coyle T.E.: Platelet transfusions and bleeding complications associated with plasma exchange catheter placement in patients with presumed thrombotic thrombocytopenic purpura. J Clin Apheresis 2013; 28: pp. 356-358.
- Brunskill S.J., Tusold A., Benjamin S., et al: A systematic review of randomized controlled trials for plasma exchange in the treatment of thrombotic thrombocytopenic purpura. Transfus Med 2007; 17: pp. 17-35.
- Shumak K.H., Rock G.A., and Nair R.C.: Late relapses in patients successfully treated for thrombotic thrombocytopenic purpura. Ann Intern Med 1995; 122: pp. 569-572.
- http://www.ncbi.nlm.nih.gov/pubmed/24929773
- http://www.ncbi.nlm.nih.gov/pubmed/25060255
- http://www.ncbi.nlm.nih.gov/pubmed/24472354





4 thoughts on “Thrombotic Thrombocytopenic Purpura: Pearls and Pitfalls”
Solid work Britt!!
Pingback: Asynchronous Learning: Hematology and Oncology - Bold City Emergency Medicine
Pingback: Best Case Ever 53 - TTP - Emergency Medicine Cases
Pingback: Tasty Morsels of EM 070 – TTP - Emergency Medicine Ireland