
The EM Educator Series: Empyema – Does this “pneumonia” need source control?
The EM Educator series looks at empyema.
The EM Educator Series: Empyema – Does this “pneumonia” need source control? Read More »
The EM Educator series looks at empyema.
The EM Educator Series: Empyema – Does this “pneumonia” need source control? Read More »

This post covers the presentation, evaluation, and management of empyema.
Empyema: ED Presentation, Evaluation, and Management Read More »
The emDOCs Podcast covers severe asthma and asthma mimics.
emDOCs Podcast – Episode 36: Severe Asthma and Mimics Read More »

What should you consider in determining disposition for the patient with asthma or COPD exacerbation?
Asthma and COPD Evidence-Based Disposition in the ED Read More »

What can you use to determine whether the patient with community acquired pneumonia can be discharged from the ED?
Evidence-Based Disposition of Community-Acquired Pneumonia Read More »
A 66-year-old male presents via EMS in respiratory distress. For the past several days he has had worsening productive cough, chest pain, dyspnea, and fever. He appears fatigued, is sitting up in bed, and has increased work of breathing. On exam there is no JVD, 1+ edema, and crackles bilaterally. Vital signs include T 102.1 F, HR 110, RR 28, BP 94/62. Chest Xray shows bilateral infiltrates. What is the likely diagnosis?
EM@3AM: Acute Respiratory Distress Syndrome Read More »
A 30-year-old male presents after a motor vehicle accident with chest pain and shortness of breath. He is alert, speaking in 1-2 word responses, and appears in moderate respiratory distress. He has normal left sided breath sounds, but none on the right. No lung sliding is seen with ultrasound examination of the right thorax. His trachea is deviated left, and increased JVD is noted. What is this patient’s presentation consistent with? What is your next step in management?
EM@3AM: Pneumothorax Read More »
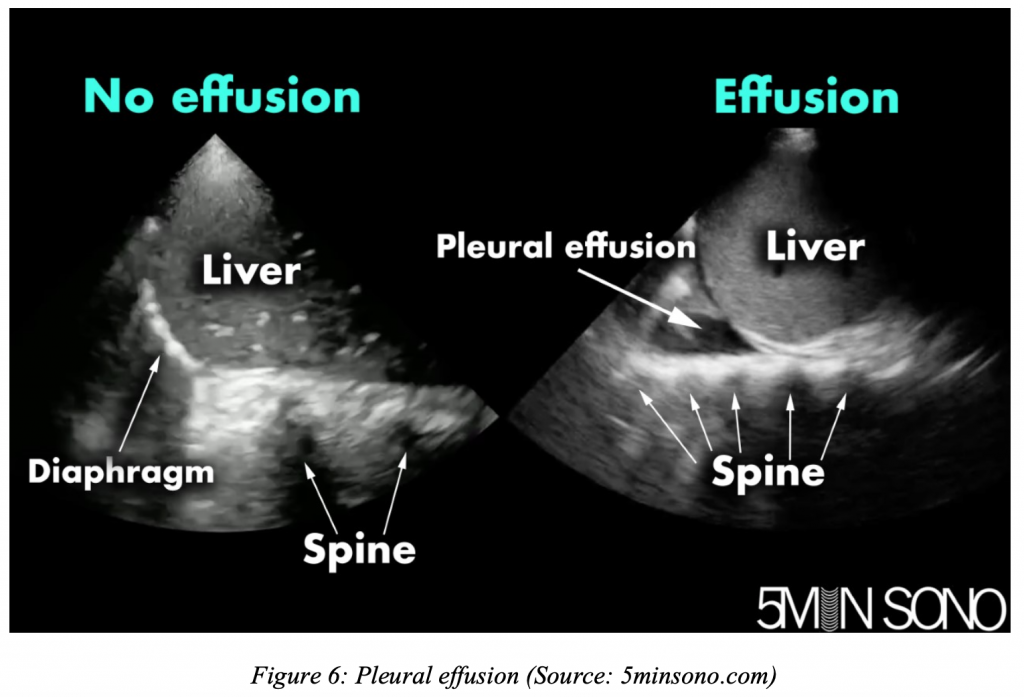
How do you evaluate for suspected pleural effusion, and what tools do you have for diagnosis and therapy?
ED evaluation and management of pleural effusions: One size doesn’t fit all Read More »

Up in Smoke about E-Cigarettes and the Pulmonary Wasteland
TOXCard: Vaping Associated Pulmonary Injury, 2019 outbreak Read More »

The last community-acquired pneumonia guidelines from the ATS/IDSA were released in 2007. Ready for an update?
Community-Acquired Pneumonia: ATS/IDSA Guidelines Update Read More »