
Authors: Daniel A. Weidner, DO (EM Resident Physician, San Antonio, TX) and Michael J. Yoo, MD (EM Resident Physician, San Antonio, TX) // Reviewed by: Alex Koyfman, MD (@EMHighAK) and Brit Long, MD (@long_brit)
Case
A 69-year-old man with a history of recurrent prostate cancer, hypertension, and diabetes presents to the emergency department (ED) with a chief complaint of lower abdominal pain. The patient states that he has been compliant with his medications but has not yet started chemotherapy or radiation therapy. He notes that he has had issues urinating for several weeks but has not been able to urinate for the past 3 days. He denies any infectious symptoms.
His vital signs are HR 104, BP 174/89, RR 20, 96% on RA, and temperature of 37 C. Exam is notable for a non-toxic appearing man with significant suprapubic pain, unremarkable genitalia, and a large multinodular prostate. What are your next steps in management?
This article will cover the approach to oliguria and anuria in the ED with presentation, evaluation, and management of common causes of reduced urine output. The authors also highlight some less common but serious etiologies of oliguria and anuria. This article will exclude pediatric specific causes and chronic anuria in dialysis patients.
Background and Definitions
While there are several definitions of oliguria and anuria, generally, oliguria is defined as urinary output of less than 0.5 mL/kg/hour over a 6 hour period or a total of 100-400 mL over a 24 hour period.1-3 With disease progression, patients can develop anuria, typically defined as urinary output of less than 100 mL over a 24 hour period.1 The ability to resorb electrolytes and water are normal physiologic functions of the kidney, but decreased urinary output can be an indication of progression to acute kidney injury (AKI). Similar to oliguria and anuria, many definitions exist. One of the most commonly used criteria is the Kidney Disease: Improving Global Outcomes (KDIGO) definition below:2
Table 1. Stages of AKI, as defined by the KDIGO criteria.2

Though oliguria/anuria and AKI have many overlapping features and causes, their definitions differ as discussed above. Not all patients with oliguria/anuria have AKI, and not all patients with acute renal injury have oliguria/anuria.
Epidemiology and Significance of Oliguria and Anuria
Up to half of all patients admitted to the intensive care unit experience oliguria at some point during their stay, with 30% of these patients having oliguria lasting at least 6 hours.3 Risk factors for developing oliguria and AKI include diabetes, hypertension, advanced age, heart failure, sepsis, and underlying CKD.4,5
Decreasing urinary output, to include oliguria and anuria, prognosticate deteriorating renal function and may be the only clue of renal insult in the early stages of AKI.6-8 While creatinine is a surrogate for kidney function, serum creatinine lags behind true kidney function and can be obscured by fluid overload states.6-7 Furthermore, progression to anuria has been associated with multiple organ dysfunction syndrome and increased risk of death compared to non-anuric AKI groups.8 Recognizing and intervening early in the course of the disease process may have the potential to improve outcomes.10,11
Differential Diagnoses and Presentation
The differential diagnosis of decreased urine output can be overwhelming, but approaching oliguria and anuria in three main categories can be helpful and include: prerenal oliguria, intrinsic oliguria, and postrenal oliguria.1,12
Table 2. Differential Diagnosis of Oliguria and Anuria in Relation to Kidneys.1,12
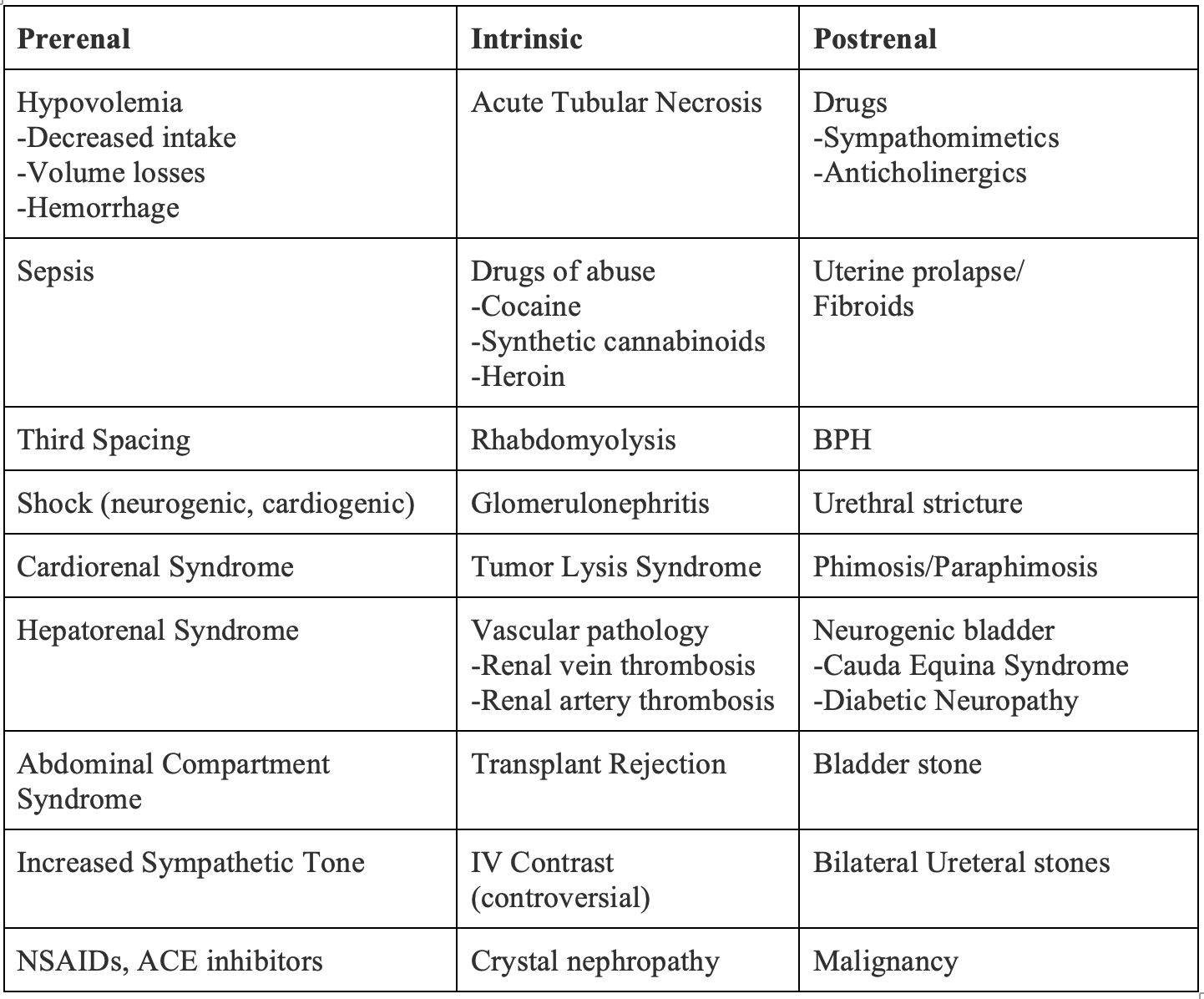
Prerenal Causes of Oliguria1,11,13
The majority of patients with oliguria have a prerenal etiology, or any disease process that reduces renal perfusion, with hypovolemia and sepsis being the most common causes.11 In a response to global hypoperfusion, both the sympathetic nervous system and renin-angiotensin-aldosterone system (RAAS) are activated, leading to direct arteriolar vasoconstriction and enhanced resorption of sodium and water. In healthy kidneys, this clinically translates to decreased urinary output. In the early stages of prerenal oliguria, the glomerular filtration rate (GFR) is maintained by a prostaglandin mediated compensatory afferent arteriolar dilation. If addressed quickly, correction of volume status and restoration of renal perfusion can prevent AKI, while persistent underperfusion can advance to ischemia. However, patients who take medications that affect the RAAS may have reduced reserve, placing them at higher risk of prerenal AKI. Notable offenders include chronic NSAID use, which inhibit prostaglandin production, and angiotensin converting enzyme (ACE) inhibitors.1,13 Clinical clues that may suggest a prerenal etiology of oliguria include signs and symptoms of hypovolemia, to include tachycardia, decreased skin turgor, dry mucous membranes, sunken eyes, or decreased capillary refill. Patients may report a history of vomiting, diarrhea, decreased oral intake, diuretic use and/or abuse, or burns with insensible losses.1,13 As mentioned above, hypovolemia and infection are typical precipitants of prerenal oliguria. Though less common, cardiorenal syndrome, hepatorenal syndrome, and abdominal compartment syndrome are important causes of prerenal oliguria highlighted below.
Figure 1. Renin-Angiotensin-Aldosterone System Physiology.

Cardiorenal Syndrome5,14
Cardiorenal syndrome is a form of prerenal dysfunction and oliguria caused by either decreased cardiac output or worsening sodium retention leading to increased renal venous congestion and decreased renal perfusion pressures. In response, the kidneys activate the RAAS, release antidiuretic hormone, and activate the sympathetic nervous system leading to further fluid retention and increased systemic vascular resistance exacerbating renal perfusion pressures. In contrast to hypovolemic etiologies of oliguria in which volume repletion is the mainstay of therapy, patients with cardiorenal syndrome are typically fluid overloaded and should be approached with optimizing cardiac output and reducing afterload with diuretics. Most common causes of acute cardiorenal syndrome include medication non-compliance, diet non-compliance, myocardial infarction, and infection. Notably, because of volume overload and subsequent bowel edema, patients with acute cardiorenal syndrome have decreased absorption of enteric diuretics, which should be avoided. A reasonable starting strategy of parenteral diuretics is to start with 1.0 to 1.5 mg/kg of intravenous furosemide and assess urine output. As a reminder, key conversions of diuretics are: 40 mg oral furosemide = 20 mg IV furosemide = 20 mg PO/IV torsemide = 1 mg PO/IV bumetanide.
Hepatorenal Syndrome15,16
Hepatorenal syndrome (HRS) is a serious complication of advanced liver disease in which worsening portal hypertension leads to splanchnic dilation and decreased effective circulatory volume. In response to decreased renal perfusion, the RAAS is activated, leading to further renal vasoconstriction, a decreased filtration rate, and oliguria. In type I HRS, patients have an acute rise in serum creatinine and urine output typically less than 500 mL/day over the course of two weeks. Type II HRS is less severe and is characterized by diuretic resistant ascites. In both types, patients with HRS have normal urinary sediment with minimal proteinuria and a low urinary sodium concentration. However, patients may be non-oliguric, especially in the early stages of HRS. Management of oliguria secondary to HRS includes addressing precipitants of acute on chronic liver injury, to include alcohol abstinence, viral hepatitis, and medication non-compliance. Patients are treated with supportive care to include vasopressors to maintain the mean arterial pressure (MAP) 10-15 mmHg above baseline and colloid infusions. This should be achieved with norepinephrine as the vasopressor of choice and albumin boluses of 1 g/kg per day. If refractory, vasopressin may be effective as an adjunct vasopressor, starting at 0.01U/min and titrating to effect. Further medical management to include the addition of midodrine (typically starting at 15 mg three times a day) or octreotide (typically starting at 50 mcg/hr) should be discussed with the intensivist or hospitalist prior to starting in the ED. Ultimately, patients who fail medical management should be evaluated for renal replacement therapy, transjugular intrahepatic portosystemic shunt placement, or liver transplantation.
Abdominal Compartment Syndrome17,18
Abdominal compartment syndrome (ACS) is a serious complication of intraabdominal hypertension (IAH) that occurs when increased intraabdominal pressure (IAP) leads to decreased abdominal perfusion pressure and end organ dysfunction. Major risk factors for ACS include diminished abdominal wall compliance, increased intraluminal visceral contents, such as ileus or obstruction, increased abdominal cavity contents (i.e. ascites, hematoma), excessive fluid resuscitation, and shock state leading to capillary leak. ACS can also lead to decreased chest wall compliance, elevated intrathoracic pressure, atelectasis, and respiratory distress. By definition, ACS occurs when IAP is greater than 20 mmHg; however oliguria can occur at much lower pressures and is often the first clinical manifestation of increasing IAP. Oliguria in ACS is a result of multiple compounding factors including compression of the IVC leading to decreased cardiac return and decreased mean arterial pressures (MAPs), compression of the renal veins leading to renal venous congestion, and formation of renal interstitial edema leading to decreased renal blood flow. Ultimately, these factors all contribute to decreased glomerular filtration and urine output. In the most severe of disease progression, exceedingly high IAPs can cause compression of the ureters and bladder leading to obstruction oliguria and anuria. The physiologic mechanism of ACS is depicted below.18
Figure 2. Pathophysiology of ACS Leading to Oliguria and Multisystem Organ Failure.18
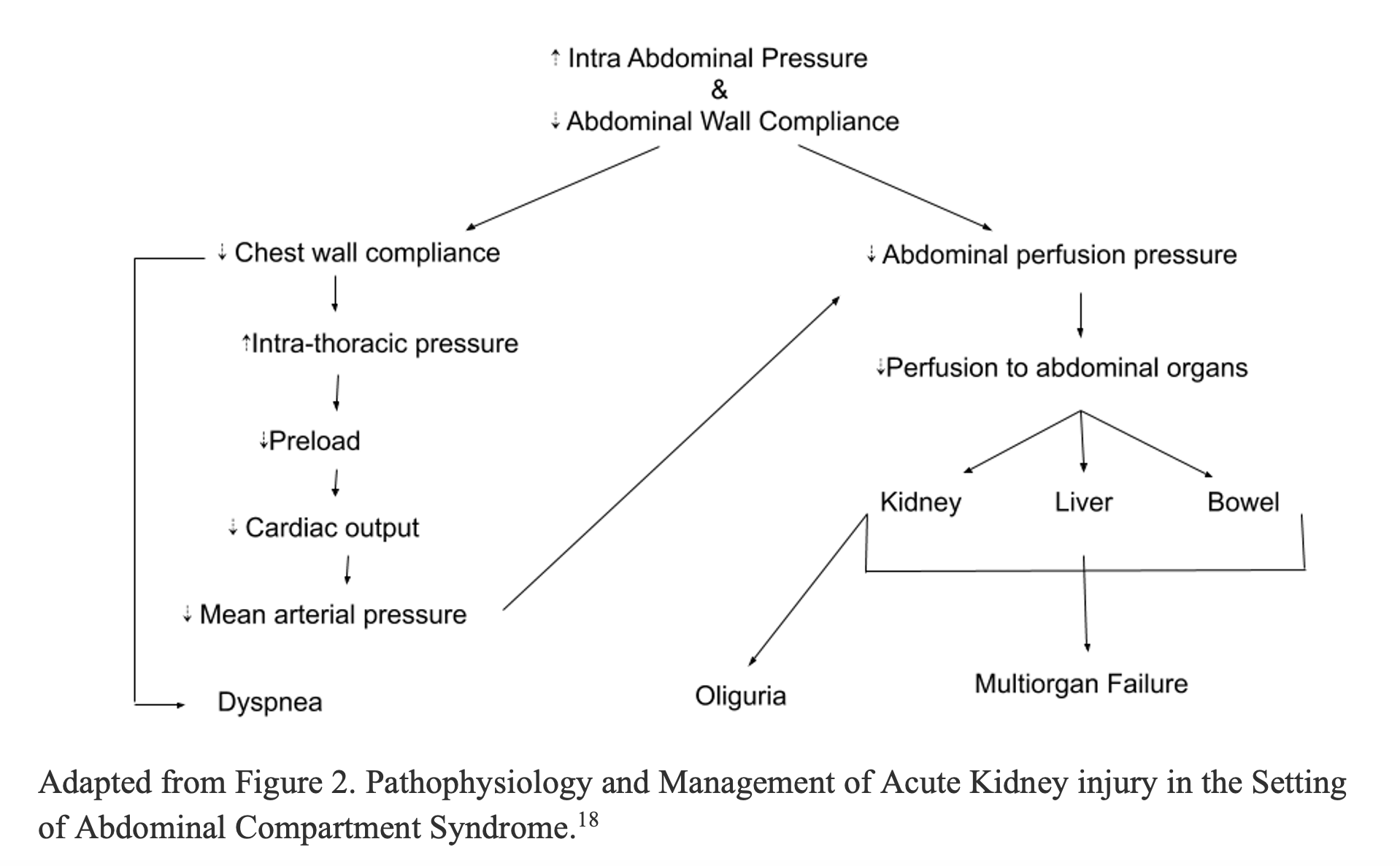
If ACS is suspected, bladder pressures are used as a surrogate for intra-abdominal pressure and can be measured via foley catheter (see link below). As a reference, normal IAP is 0-5 mmHg. While abdominal hypertension is defined as an IAP of 12-20 mmHg, an IAP greater than 20 mmHg with new onset organ dysfunction is diagnostic for ACS. The current gold standard treatment for ACS is laparoscopic decompression of the abdomen. Non-operative management strategies include: gastric decompression with a nasogastric tube, fluid removal with diuretics or paracentesis, adequate pain control, and removal of restrictive dressings.18 In cases of hypotension, vasopressors should be initiated early and fluid resuscitation used judiciously to prevent worsening of ACS. For more on abdominal compartment syndrome and foley method for measuring IAP see the following article: http://www.emdocs.net/abdominal-compartment-syndrome-pearls-pitfalls/
Sepsis19-23
Aside from hypovolemia, sepsis is the leading cause of prerenal oliguria and AKI. Up to one third of septic patients will have concomitant AKI, and oliguria is often the earliest sign of end organ dysfunction.20 Despite frequent revisions in sepsis definitions and often controversial guidelines, the pathophysiology of oliguria in sepsis remains unclear. In patients with severe sepsis, cytokines and bacterial toxins cause vasodilation, reducing the effective circulatory volume and renal perfusion pressures. Furthermore, proinflammatory molecules including pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) may have a direct effect on nephrons resulting in oxidative damage through the activation of toll-like receptors (TLRs). Both pathways likely contribute to oliguria in severe infections.20-21
We briefly review the systemic inflammatory response syndrome (SIRS) and quick sepsis related organ failure (qSOFA) guidelines below, which is discussed further in depth here: http://www.emdocs.net/em-educator-series-sepsis-ed/
Table 3. SIRS and qSOFA Criteria.19,21

The mainstay of treatment for sepsis is prompt fluid resuscitation, source control, and early broad spectrum antibiotic therapy aimed at the most likely bacterial source.21-22 While traditional guidelines suggest an initial 30 cc/kg fluid bolus for septic patients, factors to include underlying cardiac, renal, and hepatic disease prove challenging in resuscitation; moreover, fluid overloaded states may be associated with increased morbidity and mortality.24-26 In these patients, judicious fluid administration regardless of the presence or absence of oliguria and early use of vasopressors should be considered.22
Intrinsic Cause of Oliguria
In contrast to prerenal and postrenal oliguria, intrinsic causes of oliguria are due to direct parenchymal damage from toxins, ischemic, or autoimmune processes. Acute tubular necrosis (ATN) is a common cause of intrinsic oliguria and occurs when ischemia or direct damage to the renal tubule epithelium results in sloughing of the endothelial lining.27Subsequent renal tubule obstruction and hydrostatic leak of filtrate into the bloodstream can result in electrolyte derangement and oliguria.27 Muddy brown casts are classically seen on microscopic urinalysis. If there is a suspicion for an intrinsic etiology of oliguria, identifying and stopping any inciting factors, avoiding additional nephrotoxins, and hydration remain at the foundation of therapy. Rhabdomyolysis, tumor lysis syndrome, renal ischemia, acute kidney transplant rejection, and nephrotoxic medications including drugs of abuse are important causes of intrinsic oliguria. Rhabdomyolysis and a review of nephrotoxic medications and drugs of abuse are highlighted below.
Nephrotoxins13,27-28
The following medications are common nephrotoxic medications that should be reviewed in any patient with oliguria. When possible, these medications should be discontinued or avoided.
Table 4. Nephrotoxins and their Mechanism of Nephrotoxicity.

Rhabdomyolysis1,13,29
Rhabdomyolysis results from skeletal muscle breakdown and leakage of intracellular contents into the bloodstream. In addition to electrolyte derangements, to include hyperkalemia, rhabdomyolysis causes increased concentrations of myoglobin and its byproduct ferrihemate and uric acid, all of which cause direct insult to the kidneys by causing intratubular cast formation and obstruction. Classically, patients complain of tea colored urine after being exposed to crush, heat, and electrical injuries, seizure, or extreme exertion. Diagnostically, creatine kinase (CK) levels are 5 times the upper limit of normal, with levels in the ten thousands being common in more severe cases. A urinalysis that is positive for blood with negative red blood cells on microscopic analysis further supports a diagnosis of rhabdomyolysis. Patients should be aggressively hydrated, with a target urine output of 2-3 cc/kg/hr. Urinary alkalinization with sodium bicarbonate is controversial without enough research to support its universal use. For a more comprehensive review of this topic, please refer to a previous emDOCs article: http://www.emdocs.net/emdocs-cases-evidence-based-recommendations-for-rhabdomyolysis/
Post-Renal13,30
Post-renal causes of oliguria, and in severe cases, anuria, are almost exclusively caused by physical obstruction of the urinary tract, to include the ureters (bilaterally or in the case of one functioning kidney), bladder, and urethra preventing forward flow of filtrate and urine. Important clues in the history include: benign prostatic hyperplasia (BPH) and prostate cancer in men, urethral trauma, kidney stones, pelvic surgery or trauma, spine trauma, and pelvic organ prolapse.13 Anticholinergic medications can cause a pseudo-obstruction by inhibiting bladder contraction during urination.30 Of note, despite the initial oliguria, patients with an obstructive physiology may still urinate due to overflow incontinence.30 Physical exam findings suggestive of an obstructive etiology include a palpable bladder, a large prostate on rectal exam in men, pelvic organ prolapse in women, and blood at the meatus in trauma patients.13,30
With the exception of patients with concern for urethral trauma, bladder decompression with a Foley catheter is the first step in management of obstructive causes of oliguria. Foley catheters have a wide range of sizes, as large as 28 French; however, a 14 or 16 French foley catheter is a reasonable starting point. In patients with a known history of BPH or prostate cancer, starting with a Coude catheter (with a bent tip) can help navigate around the prostate and facilitate first pass success. An additional consideration includes patients with a suspicion for blood clots as an obstructive cause. In these patients, a three-way Foley can facilitate irrigation of the bladder and clearance of the blood clots. If all attempts fail, suprapubic aspiration or catheterization should be attempted to prevent bladder rupture and peritonitis. A review of this procedure can be found here: http://www.emdocs.net/suprapubic-catheter-complications-tips-tricks/
Pearl: Remember to consider cauda equina and conus medullaris syndrome on the differential for acute urinary retention. Though sensitivities are poor (50-70%), acute urinary retention can be the earliest presenting sign of patients with acute spinal compressive syndromes.30 Patients should undergo magnetic resonance imaging (MRI) of the T- and L- spine with contrast and be evaluated for neurosurgical decompression. A further discussion of cauda equina syndrome can be found here: http://www.emdocs.net/em3am-cauda-equina-syndrome/
Case resolution
You perform a bedside ultrasound and find a large bladder, with approximately 1.3L of urine on volume measurement. You place a 16 French Coude catheter with immediate relief in the patient’s symptoms. Labs are only notable for a mild elevation in creatinine not meeting AKI criteria. You arrange for him to be seen in an outpatient urology clinic and discharge the patient with the Coude catheter in place.
Take Home Points
-Oliguria has a broad differential: rule out obstruction first.
-Consider neurogenic causes of acute urinary retention such as cauda equina syndrome.
-Search for and avoid nephrotoxins if possible: NSAIDs, ACEi/ARB, antibiotics, antivirals, diuretics, and drugs of abuse.
-Prerenal etiologies are the most common cause of oliguria and are often caused by hypovolemia or infection; however, can’t miss causes include cardiorenal syndrome, hepatorenal syndrome, and abdominal compartment syndrome.
-Relieve obstructions with a Foley catheter, and consider a Coude catheter in patients with large prostates and a three-way catheter in patients with bleeding and clots.
References/Further Reading
1. Walls RM, Hockberger RS, Gausche-Hill M, Bakes KM, Wolfson AB. Chapter 87: Renal Failure. In: Rosen’s Emergency Medicine: Concepts and Clinical Practice. Philadelphia, PA: Elsevier; 2018:1179-1196.
2. Section 2: AKI Definition. Kidney Int Suppl (2011). 2012;2(1):19-36. doi:10.1038/kisup.2011.32
3. Klein SJ, Lehner GF, Forni LG, Joannidis M. Oliguria in critically ill patients: a narrative review. J Nephrol. 2018;31(6):855-862. doi:10.1007/s40620-018-0539-6
4. Pavkov ME, Harding JL, Burrows NR. Trends in Hospitalizations for Acute Kidney Injury – United States, 2000-2014. MMWR Morb Mortal Wkly Rep. 2018;67(10):289-293. Published 2018 Mar 16. doi:10.15585/mmwr.mm6710a2
5. Jentzer JC, Bihorac A, Brusca SB, et al. Contemporary Management of Severe Acute Kidney Injury and Refractory Cardiorenal Syndrome: JACC Council Perspectives. J Am Coll Cardiol. 2020;76(9):1084-1101. doi:10.1016/j.jacc.2020.06.070
6. Vincent JL, Ferguson A, Pickkers P, et al. The clinical relevance of oliguria in the critically ill patient: analysis of a large observational database. Crit Care. 2020;24(1):171. Published 2020 Apr 23. doi:10.1186/s13054-020-02858-x
7. Kellum JA, Sileanu FE, Murugan R, Lucko N, Shaw AD, Clermont G. Classifying AKI by urine output versus serum creatinine level. J Am Soc Nephrol. 2015;26(9):2231-2238. doi:10.1681/ASN.2014070724
8. Macedo E, Malhotra R, Bouchard J, Wynn SK, Mehta RL. Oliguria is an early predictor of higher mortality in critically ill patients. Kidney Int. 2011;80(7):760-767. doi:10.1038/ki.2011.150
9. Choi HM, Kim SC, Kim MG, Jo SK, Cho WY, Kim HK. Etiology and outcomes of anuria in acute kidney injury: A single center study. Kidney Res Clin Pract. 2015;34(1):13-19. doi:10.1016/j.krcp.2014.11.002
10. Koeze J, Keus F, Dieperink W, Van der Horst ICC, Zijlstra JG, Van Meurs M. Incidence, timing and outcome of AKI in critically ill patients varies with the definition used and the addition of urine output criteria. BMC Nephrol. 2017;18(1):1-9. doi:10.1186/s12882-017-0487-8
11. Perner A, Prowle J, Joannidis M, Young P, Hjortrup PB, Pettilä V. Fluid management in acute kidney injury. Intensive Care Med. 2017;43(6):807-815. doi:10.1007/s00134-017-4817-x
12. Gaibi T, Ghatak-Roy A. Approach to Acute Kidney Injuries in the Emergency Department. Emerg Med Clin North Am. 2019;37(4):661-677. doi:10.1016/j.emc.2019.07.006
13. Sinert R, Peacock Jr. PR. Acute Kidney Injury. In: Tintinalli JE, Ma O, Yealy DM, Meckler GD, Stapczynski J, Cline DM, Thomas SH. eds. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide, 9e. McGraw-Hill; Accessed November 25, 2020. https://accessmedicine.mhmedical.com/content.aspx?bookid=2353§ionid=219671573
14. Jentzer JC, Chawla LS. A Clinical Approach to the Acute Cardiorenal Syndrome. Crit Care Clin. 2015;31(4):685-703. doi:10.1016/j.ccc.2015.06.006
15. Tapper EB, Bonder A, Cardenas A. Preventing and Treating Acute Kidney Injury among Hospitalized Patients with Cirrhosis and Ascites: A Narrative Review. Am J Med. 2016;129(5):461-467. doi:10.1016/j.amjmed.2015.12.006
16. Crager S. Critically Ill Patients with End-Stage Liver Disease. Emerg Med Clin North Am. 2019;37(3):511-527. doi:10.1016/j.emc.2019.03.008
17. Patel DM, Connor MJ. Intra-Abdominal Hypertension and Abdominal Compartment Syndrome: An Underappreciated Cause of Acute Kidney Injury. Adv Chronic Kidney Dis. 2016;23(3):160-166. doi:10.1053/j.ackd.2016.03.002
18. Sandhu G, Mankal P, Gupta I, Ranade A, Bansal A, Jones J. Pathophysiology and management of acute kidney injury in the setting of abdominal compartment syndrome. Am J Ther. 2014;21(3):211-216. doi:10.1097/MJT.0b013e318235f1cf
19. Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801-810. doi:10.1001/jama.2016.0287
20. Peerapornratana S, Manrique-Caballero CL, Gómez H, Kellum JA. Acute kidney injury from sepsis: current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int. 2019;96(5):1083-1099. doi:10.1016/j.kint.2019.05.026
21. Puskarich MA, Jones AE. Sepsis. In: Tintinalli JE, Ma O, Yealy DM, Meckler GD, Stapczynski J, Cline DM, Thomas SH. eds. Tintinalli’s Emergency Medicine: A Comprehensive Study Guide, 9e. McGraw-Hill; Accessed December 08, 2020. https://accessemergencymedicine.mhmedical.com/content.aspx?bookid=2353§ionid=220292051
22. Poston JT, Koyner JL. Sepsis associated acute kidney injury. BMJ. 2019;364:k4891. Published 2019 Jan 9. doi:10.1136/bmj.k4891
23. Levy, M.M., Evans, L.E. & Rhodes, A. The Surviving Sepsis Campaign Bundle: 2018 update. Intensive Care Med 44, 925–928 (2018). doi.org/10.1007/s00134-018-5085-0
24. Mehta, R. Fluid balance and acute kidney injury: the missing link for predicting adverse outcomes? Nat Rev Nephrol5, 10–11 (2009). doi.org/10.1038/ncpneph0988
25. Wang N, Jiang L, Zhu B, Wen Y, Xi XM. Fluid balance and mortality in critically ill patients with acute kidney injury: A multicenter prospective epidemiological study. Crit Care. 2015;19(1):1-11. doi:10.1186/s13054-015-1085-4
26. Bellomo R, Kellum JA, Ronco C, et al. Acute kidney injury in sepsis. Intensive Care Med. 2017;43(6):816-828. doi:10.1007/s00134-017-4755-7
27. Ghannoum M, Goldfarb DS. Renal Principles. In: Nelson LS, Howland M, Lewin NA, Smith SW, Goldfrank LR, Hoffman RS. eds. Goldfrank’s Toxicologic Emergencies, 11e. McGraw-Hill; Accessed November 04, 2020.
28. Mansoor K, Kheetan M, Shahnawaz S, et al. Systematic review of nephrotoxicity of drugs of abuse, 2005-2016. BMC Nephrol. 2017;18(1). doi:10.1186/s12882-017-0794-0
29. Chavez LO, Leon M, Einav S, Varon J. Beyond muscle destruction: a systematic review of rhabdomyolysis for clinical practice. Crit Care. 2016;20(1):135. Published 2016 Jun 15. doi:10.1186/s13054-016-1314-5
30. Billet M, Windsor TA. Urinary Retention. Emerg Med Clin North Am. 2019;37(4):649-660. doi:10.1016/j.emc.2019.07.005




