Authors: Colton Margus, MD (@ColtonMargus, EM Resident Physician, Mount Sinai St. Luke’s-Roosevelt) and Jenny Beck-Esmay, MD (@jbeckesmay, Assistant Residency Program Director, Mount Sinai St. Luke’s-Roosevelt) // Edited by: Alex Koyfman, MD (@EMHighAK); Tim Montrief, MD (@EMinMiami); Brit Long, MD (@long_brit)
Case: A 65-year-old woman with a history of alcoholic cirrhosis comes to the emergency department from her nursing home for altered mental status. There’s a report that suggests she was recently admitted to an outside hospital for hematemesis and confusion, but no endoscopy is documented. Nursing home staff endorsed only a vague, progressive confusion and decreased urine output. Per EMS, the patient was found lying awake in bed, confused but speaking. However, upon arrival in the emergency department, the patient appeared jaundiced with significant scleral icterus and would only open her eyes to painful stimuli, without purposeful movement. Her triage vital signs were a blood pressure of 89/52 mm Hg, heart rate of 93 beats per minute, respiratory rate of 21 breaths per minute, oxygen saturation of 99% on room air, and a core temperature of 35.1 °C. What is on your differential for this patient? What are your next steps?
Background
Liver cirrhosis is a disease with multisystem sequelae, as shown by this case presentation of likely hepatic encephalopathy with a recent history of variceal bleed. But, for this patient, the renal manifestations of a cirrhotic circulatory system are what ultimately prove of greatest consequence. First described in 1863, hepatorenal syndrome (HRS) is, by definition, a type of oliguric renal failure due to liver disease in the absence of any intrinsic renal pathology.1 As portal venous congestion develops in the setting of cirrhosis, renal hypoperfusion leads to kidney injury and precipitous decline in renal function. One follow-up investigation of 234 non-azotemic patients with cirrhosis found that the probability of HRS at 1 year was 18% and, at 5 years, approached 39%.2 This syndrome is sometimes further differentiated into type 1 and 2 depending on whether it develops rapidly as an acute kidney injury (≥100% rise in creatinine in less than two weeks) or more insidiously as a chronic ascites-associated disease, respectively.3 In either case, HRS represents the end-stage liver failure.
Pathophysiology
The pathogenesis of HRS is incompletely understood but is as much about the vasculature as it is about the liver and kidneys. As cirrhosis progressively impedes portal venous flow, it causes backup into the splanchnic circulation (the celiac, superior mesenteric, and inferior mesenteric offshoots of the abdominal aorta). While the splanchnic circulation tries to counter this strain with local release of vasodilators including nitric oxide,4the systemic circulation reacts to this vasodilation with its own counter-response of systemic vasoconstrictors, activating the sympathetic and renin-angiotensin-aldosterone (RAS) systems. Together, these have a net effect of clamping down on renal blood flow. For example, the classical RAS pathway increases circulating angiotensin II levels and thereby directly stimulates renal vasoconstriction in these patients.5
In other words, blood flow through the liver is impeded, causing vasodilators to accumulate upstream and a broadly hyperdynamic, hyporesponsive vasculature. The body tries to counter this with its usual autonomic and volume-retention measures, and kidney function deteriorates as a result.
There is still much unknown about the pathogenesis of HRS. For example, a recent study measuring inflammatory markers in 522 patients with acute-on-chronic liver failure found the degree of renal failure to be associated with inflammatory markers rather than plasma renin, as would have been expected if RAS activation were the whole picture.6 Clearly, the true complexity behind the syndrome remains to be elucidated.
Presentation
HRS is a late consequence of liver failure and therefore often presents with stigmata of acute-on-chronic liver failure. Patients may present with significant ascites and peripheral edema, jaundice, physical manifestations of coagulopathy, and/or hepatic encephalopathy.7 Oliguria is not always immediately apparent and may or may not be seen in the emergency department, depending on the timing of evaluation and diagnosis.7 Traditionally, HRS is subdivided based on onset: Type I that is more acute, often with a precipitating event, and type II that is more gradual and with more substantial and resistant ascites.8

Regardless of urine output, the failure of the kidneys to properly excrete solute-free water manifests as hyponatremia, third-spacing, and hypotension.7 Attention should also be paid to predisposing factors, such as gastro-intestinal bleeding, cholestasis, and significant ascites or peritonitis.9 Evidence of fever and other signs or symptoms of infection are highly concerning, as infection is associated with significant mortality risk in these patients.3
The differential for kidney injury in cirrhotic patients is broad, with HRS considered a diagnosis of exclusion.3 Other pathologies to consider include pre-renal acute kidney injury, infection (for example, pyelonephritis, sepsis, or spontaneous bacterial peritonitis), or intrinsic renal diseases such as glomerulonephritides.10
Diagnostic Testing
In any patient with signs and symptoms of progressive liver failure, it is important to evaluate for liver function, as well as for evidence of end-organ dysfunction. Laboratory tests should include a complete blood count, metabolic panel, blood gas, lactate, ammonia, and coagulation studies, in addition to the hepatic panel of bilirubin, protein, albumin, and transaminases.2 Urine evaluation should include urinalysis, urine osmolality, as well as calculation of a fractional sodium excretion (FENa) and fractional urea excretion (FEUrea).11
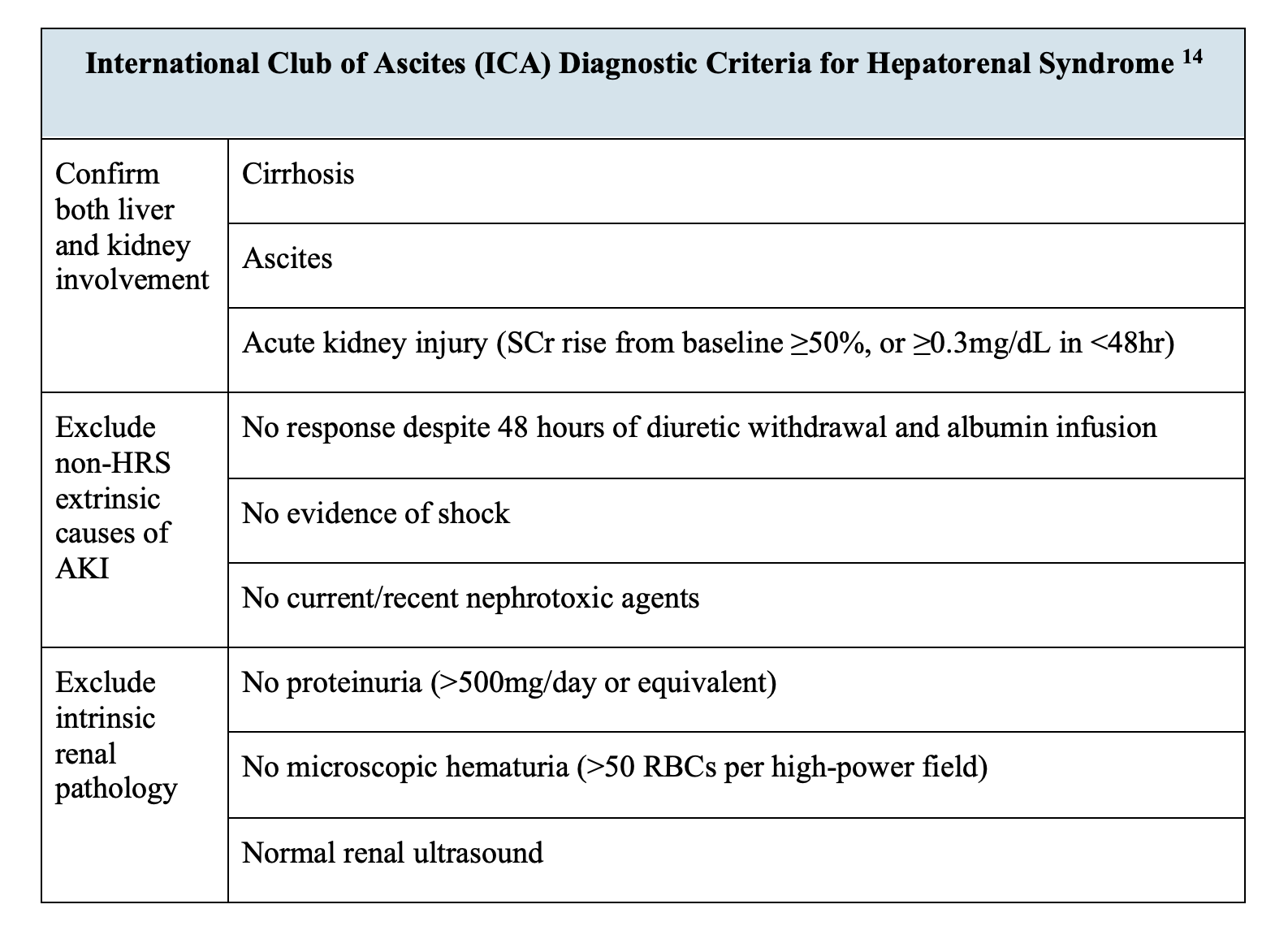
How will these labs help? The first step in the diagnosis of HRS in a patient with liver failure is to recognize acute kidney injury (AKI). Serum creatinine (sCr) not only varies with sex, age, and BMI but also with severe hepatic dysfunction. Such dysfunction impedes creatine to creatinine conversion, while also decreasing muscle mass for creatine supply in the first place.3 AKI in these patients is diagnosed per the International Club of Ascites guidelines if there is an increase in sCr ≥50% from baseline or a ≥0.3mg/dL increase from initial sCr within 48 hours of admission.12
From there, the AKI must still be further differentiated, as HRS should be thought of as a diagnosis of exclusion.3 HRS is far from the most common cause of kidney injury in cirrhotic patients: as one prospective study of 562 cirrhotic patients found, renal failure in this population was most commonly due to spontaneous bacterial peritonitis (SBP, 46%), followed by hypovolemia (32%) and then finally HRS (13%).13 Therefore, in a patient with cirrhosis, ascites, and AKI, ruling out other causes of the kidney injury through diagnostic and potentially therapeutic interventions precede the definitive diagnosis of HRS. These include stopping any nephrotoxic, vasodilatory, and diuretic drugs and treating hypovolemia as well as any underlying infections.11
Another key criterion for diagnosing HRS is the lack of response to volume replacement with 1g/kg/day albumin (not exceeding 100g/day) over two days.14 Because this won’t be determined in the ED, it remains important for the emergency physician to continue to consider other etiologies. For example, hydronephrosis on renal ultrasound might indicate an obstructive uropathy.14 Protein with red blood cells might indicate glomerulonephritis (especially in the setting of hepatitis), while white blood cell or granular casts might indicate interstitial nephritis or acute tubular necrosis, respectively.15 Of course, these delineations are frequently blurred: a severe drop in renal blood flow in HRS can lead to tubular necrosis, bile cast formation and resulting tubular obstruction.16
Even in a patient with the expected high urinary osmolality but low FENa and no urinary blood or protein,17 there remain other considerations to rule out before diagnosing HRS, such as prerenal azotemia and abdominal compartment syndrome.18 One recent retrospective cohort analysis of cirrhotic patients with ascites and AKI found a fractional excretion of urea less than 28% to be 75% sensitive and 83% specific for HRS.19
Management
HRS is an indirect consequence of splanchnic vasodilation and relative hypovolemia. Because of this, systemic vasoconstrictors are the current first line therapy for HRS, as an attempt at thwarting the body’s own compensatory mechanisms that restrict renal perfusion.20 Terlipressin (at 0.5-1mg IV every 4 to 6 hours) is one drug of choice but not readily available in the United States.21 Lack of FDA approval may be due to case reports of ischemic events, such as skin necrosis, observed with terlipressin use.22,23 One alternative therapy to terlipressin is the somatostatin analogue octreotide (100-200mcg SQ every 8 hours) plus alpha-1 agonist midodrine (7.5-12.5mg orally three times daily). Norepinephrine (0.5-3mg per hour IV infusion) may also be used as an alternative to these treatments.22
There is not much data available comparing and contrasting these three alternatives of terlipressin, octreotide with midodrine, and norepinephrine. However, a 2017 systematic review and network meta-analysis of 13 randomized control trials involving 739 patients with type 1 HRS supported the use of terlipressin (OR 26.25, 95% CI 3.07-224.21) or norepinephrine (OR 10.00, 95% CI 1.49-50.00) over midodrine with octreotide in decreasing short-term (4 week) mortality.20 Given U.S. availability and fewer adverse events, norepinephrine is probably the best approach in the emergency department setting.24 And, while there are no publications available to offer a clear blood pressure target, the Acute Dialysis Quality Initiative (ADQI) and a 2016 expert statement suggest an increase in mean arterial blood pressure (MAP) of 10 mmHg above the patient’s baseline, if known (this recommendation is based on a cohort of patients with MAP of 74mmHg).25,26
Regardless of the choice of vasoconstrictor, remember that a classic finding in cirrhotics is hypoalbuminemia, so intravenous albumin supplementation should be initiated as part of early management at 1g/kg/day.27 Such albumin infusion comes broadly recommended, but there is increasing evidence for significant hemodynamic heterogeneity in HRS, suggesting that patients might be better served by ultrasound-guided tailoring of treatment based on volume status.28 Monitor for pulmonary edema, which would warrant reduction or even cessation of albumin therapy.29
Should these treatments with colloid and vasoconstrictor fail, inpatient options for resistant HRS include renal replacement therapy (RRT), transjugular intrahepatic porto-systemic shunt (TIPS), and, as definitive treatment, liver transplantation.3 Despite no proven survival advantage, RRT becomes a final option when a patient who is failing traditional therapies and who is unable to undergo transplantation begins to succumb to metabolic derangement (acidosis, hyperkalemia), hepatic encephalopathy, and/or volume overload.25
Again, it is worth reiterating that HRS is not the most common cause of renal failure in cirrhotics. In particular, two other complications of cirrhosis, abdominal compartment syndrome and SBP are both worth mentioning. Renal dysfunction is one of the earliest signs that intra-abdominal hypertension is reaching the point of reduced organ perfusion (>20mmHg),30 and it has been shown that paracentesis can improve creatinine clearance in cirrhotic patients with extensive ascites.31 Unfortunately, although intra-abdominal pressure can be quite accurately estimated in the ED through urinary catheter-derived bladder pressures,32 how and when therapeutic paracentesis can best be used to improve kidney function in patients with cirrhosis and ascites remains unclear.30 This is a particularly challenging question given that large-volume paracentesis is a known precipitator of HRS.3 Diagnostic paracentesis, though, is of significant value: a superimposed bacterial infection of the bloodstream, lungs, or ascitic fluid can be difficult to manage and lead to sepsis and rapid deterioration. As the incidence of SBP in hospitalized cirrhotic patients is already estimated at 12-26%, empiricallyinitiating a third-generation cephalosporin after collecting ascitic fluid, especially if the patient exhibits fever, abdominal tenderness, or altered mental status, is well within reason.24,33
Prognosis
Cirrhotic patients admitted for known or suspected HRS are in need of multidisciplinary critical care in the intensive care unit setting, given their high risk for multiorgan dysfunction and rapid decompensation.24 Despite existing treatment modalities, however, HRS is a complication of liver failure with a very poor prognosis: diagnosis with type 1 HRS confers a median survival of one month, while type 2 has a median survival of less than seven months.3 This is starkly different from the prognosis of other causes of renal failure in cirrhotics: a prospective study of 562 patients demonstrated 3-month survival for parenchymal nephropathy, hypovolemic renal failure, infection-associated renal failure, and HRS of 73%, 46%, 31%, and 15% respectively.10 As a predictor of short-term mortality, the new Model for End-Stage Liver Disease-Sodium (MELD-Na score) should be used in place of traditional scoring systems, as the inclusion of serum sodium concentration incorporates the hyponatremia and impaired solute-free water excretion attributed to HRS and, by extension, increased mortality.13
Case Conclusion: In the ED, the patient was fluid resuscitated with normal saline and started on a third-generation cephalosporin empirically. Ultimately, she required intubation for airway protection and was admitted for hepatic encephalopathy and renal failure, with a serum creatinine of 2.8mg/dL. Despite the ICU team starting her on intravenous albumin, lactulose, rifaximin, and midodrine, the patient continued to decline over the subsequent days. She was never extubated, becoming increasingly unstable and expiring 12 days into her hospital stay.
Pearls & Pitfalls
-HRSis a high mortality complication of cirrhosis (1-7 month prognosis).
-HRS likely develops when portal hypertension leads to release of excess vasodilators, thereby triggering a compensatory restriction to kidney perfusion and function.
-HRS should be considered in all patients with cirrhosis + ascites + AKI refractory to volume resuscitation.
-HRS is a diagnosis of exclusion, after diuretics and nephrotoxic agents have been stopped and shock and intrinsic renal pathology ruled out.
–HRS evaluation is aided by ultrasound: to assess volume status, rule out obstructive nephropathy, and assist with paracentesis so as to exclude another big complication of cirrhosis, spontaneous bacterial peritonitis.
-HRS is a cirrhotic AKI that can be diagnosed and treated with intravenous albumin supplementation at 1g/kg/day.
-HRS vasoconstrictor therapy can be started in the ED with norepinephrine (0.5-3mg per hour IV infusion), but octreotide+ midodrine (octreotide 100-200mcg SQ every 8 hours; midodrine 7.5-12.5mg orally three times daily) is also an option, aiming for a MAP of 10 mmHg greater than the patient’s baseline.
-HRS doesn’t exclude other complications of cirrhosis like SBP and abdominal compartment syndrome, and paracentesis should be considered with a low threshold to start empiric antibiotics (typically a third-generation cephalosporin).
Further Reading:
- Acute Liver Failure: Evidence-Based Evaluation and Management
- Hepatic Encephalopathy: Common Precipitants, Sneaky Precipitants, and Clinical Pearls
- Approach to the Sick Cirrhotic Patient
- Chronic Liver Disease and Hemostasis
- Abdominal Compartment Syndrome: Pearls & Pitfalls
- Spontaneous Bacterial Peritonitis – Pearls & Pitfalls
- EM@3AM – Acute Kidney Injury
Reference/Further Reading:
- Flint A. Clinical report on hydroperitoneum based on analysis of 46 cases. Am J Med Sci. 1863;45:306–9.
- Ginès A, et al. Incidence, predictive factors, and prognosis of the hepatorenal syndrome in cirrhosis with ascites. Gastroenterology. 1993 Jul;105(1):229-36.
- Mindikoglu AL, Pappas SC. New Developments in Hepatorenal Syndrome. Clin Gastroenterol Hepatol. 2018 Feb;16(2):162–177.e1.
- Martin PY1, Ginès P, Schrier RW. Nitric oxide as a mediator of hemodynamic abnormalities and sodium and water retention in cirrhosis. N Engl J Med. 1998 Aug 20;339(8):533-41.
- Bernardi M, Trevisani F, Gasbarrini A, Gasbarrini G. Hepatorenal disorders: role of the renin-angiotensin-aldosterone system. Semin Liver Dis. 1994 Feb;14(1):23-34.
- Clària J, Stauber RE, Coenraad MJ, et al. Systemic inflammation in decompensated cirrhosis: Characterization and role in acute-on-chronic liver failure. Hepatology. 2016 Oct;64(4):1249-64.
- Ginès P, Guevara M, Arroyo V, Rodés J. Hepatorenal syndrome. Lancet 2003; 362:1819.
- Baraldi O, Valentini C, Donati G, et al. Hepatorenal syndrome: Update on diagnosis and treatment. World J Nephrol. 2015 Nov 6;4(5):511–520.
- Meltzer J, Brentjens TE. Renal failure in patients with cirrhosis: hepatorenal syndrome and renal support strategies. Curr Opin Anaesthesiol. 2010 Apr;23(2):139-44.
- Martín-Llahí M, Guevara M, Torre A, et al. Prognostic importance of the cause of renal failure in patients with cirrhosis. Gastroenterology. 2011 Feb;140(2):488-496.
- Chancharoenthana W, Leelahavanichkul A. Acute kidney injury spectrum in patients with chronic liver disease: Where do we stand? World J Gastroenterol. 2019 Jul 28;25(28):3684–3703.
- Mehta RL, et al. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care. 2007;11(2):R31.
- Nazal L and Andrés Cárdenas A. Prognostic Markers in Patients with Ascites and Hepatorenal Syndrome. Dis Markers. 2011;31(3):139–146.
- Angeli P, et al. Diagnosis and management of acute kidney injury in patients with cirrhosis: revised consensus recommendations of the International Club of Ascites. Gut. 2015 Apr;64(4):531-7.
- Venkat D, Venkat KK. South Med J. Hepatorenal syndrome. 2010 Jul;103(7):654-61.
- Aniort J, et al. Bile Cast Nephropathy Caused by Obstructive Cholestasis. Am J Kidney Dis. 2017 Jan;69(1):143-146.
- Arroyo V, Terra C, Gines P. Advances in the pathogenesis and treatment of type-1 and type-2 hepatorenal syndrome. J Hepatol 2007;46:935-946.
- Bailey J, Shapiro MJ. Abdominal compartment syndrome. Crit Care 2000;4:23-29.
- Patidar KR, Kang L, Bajaj JS, et al. Fractional excretion of urea: A simple tool for the differential diagnosis of acute kidney injury in cirrhosis. Hepatology. 2018 Jul;68(1):224-233.
- Facciorusso A, Chandar AK, Murad MH, et al. Comparative efficacy of pharmacological strategies for management of type 1 hepatorenal syndrome: a systematic review and network meta-analysis. Lancet Gastroenterol Hepatol. 2017 Feb;2(2):94-102.
- Gifford FJ, Morling JR, Fallowfield JA. Systematic review with meta-analysis: vasoactive drugs for the treatment of hepatorenal syndrome type 1. Aliment Pharmacol Ther. 2017 Mar; 45(5):593-603.
- Zhang JQ, Zhou XM, Zhao HT, et al. Adverse events of terlipressin in liver cirrhosis with acute gastrointestinal bleeding: A clinical pharmacist’s real-world observational study. Dig Med Res. 2018;1:2.
- Chiang CW, Lin YJ, Huang YB. Terlipressin-Induced Peripheral Cyanosis in a Patient with Liver Cirrhosis and Hepatorenal Syndrome. Am J Case Rep. 2019 Jan 2;20:5-9.
- Crager S. Critically Ill Patients with End-Stage Liver Disease. Emerg Med Clin North Am. 2019 Aug;37(3):511-527.
- Nadim MK, Durand F, Kellum JA, et al. Management of the critically ill patient with cirrhosis: A multidisciplinary perspective. J Hepatol. 2016 Mar;64(3):717-35.
- Cai CX, Maddukuri G, Jaipaul N, et al. A Treat-to-Target Concept to Guide the Medical Management of Hepatorenal Syndrome. Dig Dis Sci. 2015 May;60(5):1474-81. doi: 10.1007/s10620-014-3483-x. Epub 2014 Dec 23.
- Runyon BA. Hepatology. Introduction to the revised American Association for the Study of Liver Diseases Practice Guideline management of adult patients with ascites due to cirrhosis 2012. 2013 Apr;57(4):1651-3.
- Velez JCQ, Petkovich B, Karakala N, Huggins JT. Point-of-Care Echocardiography Unveils Misclassification of Acute Kidney Injury as Hepatorenal Syndrome. Am J Nephrol. 2019 Aug 8:1-8.
- Piano S, Tonon M, Angeli P. Management of ascites and hepatorenal syndrome. Hepatol Int. 2018 Feb;12(Suppl 1):122-134.
- Mohmand H, Goldfarb S. Renal dysfunction associated with intra-abdominal hypertension and the abdominal compartment syndrome. J Am Soc Nephrol. 2011 Apr;22(4):615-21.
- Cade R, Wagemaker H, Vogel S, et al. Hepatorenal syndrome. Studies of the effect of vascular volume and intraperitoneal pressure on renal and hepatic function. Am J Med. 1987 Mar;82(3):427-38.
- Cheatham ML, Malbrain ML, Kirkpatrick A, et al. Results from the International Conference of Experts on Intra-abdominal Hypertension and Abdominal Compartment Syndrome. II. Recommendations. Intensive Care Med. 2007 Jun;33(6):951-62.
- Navasa M, Follo A, Llovet JM, et al. Randomized, comparative study of oral ofloxacin versus intravenous cefotaxime in spontaneous bacterial peritonitis. Gastroenterology. 1996 Oct;111(4):1011-7.
- Kamimura H, et al. A Case of Hepatorenal Syndrome and Abdominal Compartment Syndrome with High Renal Congestion. Am J Case Rep. 2017;18:1000–1004.
- Amin AA, Alabsawy EI, Jalan R3, Davenport A4. Epidemiology, Pathophysiology, and Management of Hepatorenal Syndrome. Semin Nephrol. 2019 Jan;39(1):17-30.
- Egerod Israelsen M, Gluud LL, Krag A. Acute kidney injury and hepatorenal syndrome in cirrhosis. J Gastroenterol Hepatol. 2015 Feb;30(2):236-43.
- Bashir MH, Iqbal S, Miller R, et al. Management and outcomes of hepatorenal syndrome at an urban academic medical center: a retrospective study. Eur J Gastroenterol Hepatol. 2019 Jun 4. doi:10.1097/MEG.0000000000001462. [Epub ahead of print].
- Huggins JT, Doelken P, Walters C, Rockey DC. Point-of-Care Echocardiography Improves Assessment of Volume Status in Cirrhosis and Hepatorenal Syndrome. Am J Med Sci. 2016 May;351(5):550-3.
- Varajic B, Cavallazzi R, Mann J, et al. High versus low mean arterial pressures in hepatorenal syndrome: A randomized controlled pilot trial. J Crit Care. 2019 Aug;52:186-192.





2 thoughts on “Hepatorenal syndrome: ED presentation, evaluation, and management”
Great article! Question | our HPB specialists tell me that it is common practice NOT to perform therapeutic large volum paracentesis if suspected SBP is present. Diagnostic paracentesis recommended first for WCC measurement
In my simple ED mindset, I see a large volume tense ascites in a sick patient and I think – let’s use US to place a drain and cover with cephalosporins
I’m sure it is more nuanced than this – what do you recommend?
Thanks
Looking at the guidelines from the American Association for the Study of Liver Disease it seems they don’t recommend using a large volume paracentesis as the default treatment for ALL ascites.
https://www.aasld.org/sites/default/files/2019-06/141020_Guideline_Ascites_4UFb_2015.pdf
Reason being, the preferred acute treatment, and maintenance treatment, is a regimen of diuretics and sodium restriction and since these meds require titration, they need to be able to experiment with them to get the right dose.
HOWEVER, they do say that ”An initial therapeutic abdominal paracentesis should be performed in patients with tense ascites. Sodium restriction and oral diuretics should then be initiated.”
If I had to guess their thought process, I would think they don’t want us routinely performing large volume taps as it interferes with their ability to properly manage most of these patients. The diagnostic para lets them know if SBP is present and then much of the ascites is managed medically? I wonder what your specialists would give as a reason? Would welcome thoughts from the hematologists out there.
But, in a true tense ascites, in a patient that is uncomfortable, a large tap may be in order and has been shown to be pretty safe. It’s certainly something I have done, and will likely continue to do, in the really tense, really uncomfortable patient or in a patient who has so much tension I will have a fluid leak if I don’t get some fluid out.
http://www.emdocs.net/unlocking-common-ed-procedures-pocket-full-of-sunshine-paracentesis-in-the-ed/
It’s certainly something I have done, and will likely continue to do, in the really tense, really uncomfortable patient or in a patient who has so much tension I will have a fluid leak if I don’t get some fluid out.
In that case, <5 liters (the cut off for a large volume para and when you should be considering albumin) is usually more than enough to relieve pressure.