
Author: Brit Long, MD (@long_brit, EM Chief Resident at SAUSHEC, USAF) // Edited by: Jennifer Robertson, MD and Alex Koyfman, MD (@EMHighAK, EM Attending Physician, UT Southwestern Medical Center / Parkland Memorial Hospital)
Case
It is 1am, and emergency medical services (EMS) calls and states that a 45-year-old male with massive hematemesis is being transported to the emergency department (ED). He is unstable per report. EMS has achieved peripheral intravenous (IV) access.
Since the patient is reported to be unstable, you begin to prepare the resuscitation room. The patient arrives and he appears very ill. You immediately assess the patient’s airway, breathing and circulation (ABCs). A nurse establishes a second IV line as soon as the patient is transferred to the ED bed.
The patient appears cachectic with a protuberant abdomen. Streaks of red are present around his oropharynx. Initial vital signs (VS) include a respiratory rate (RR) of 32, blood pressure (BP) of 72/48, heart rate (HR) of 115, temperature (T) of 37.8 Celsius, and oxygen saturation (SpO2) of 90% on 2 liters (L) nasal cannula (NC). The patient has an altered mental status and is weakly attempting to swat the nurses and paramedics away.
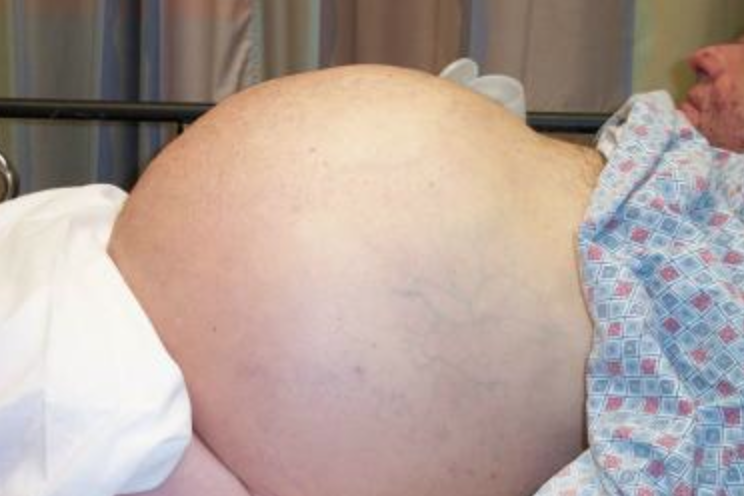
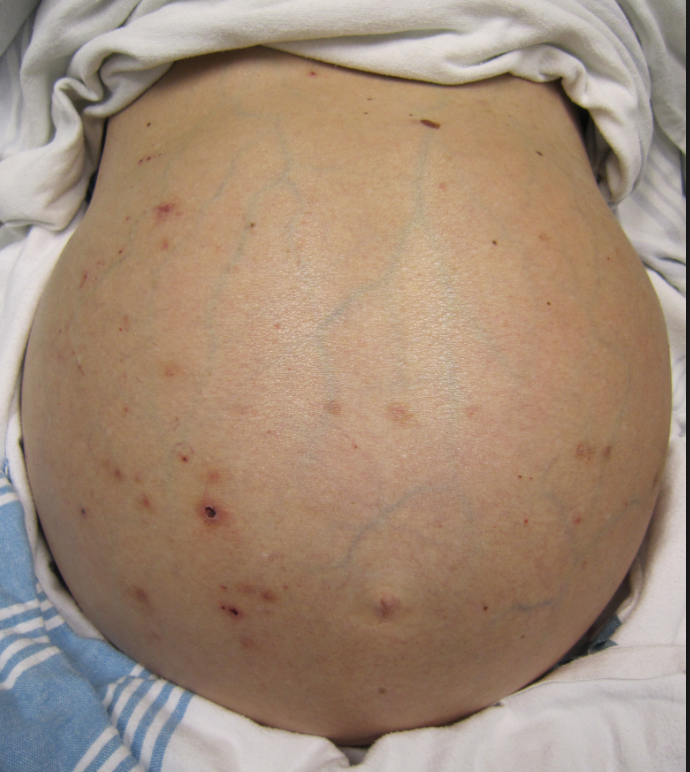
How will you approach this sick, unstable cirrhotic patient? What are some of the complications of patients with cirrhosis and chronic liver disease?
Background
Cirrhosis is the late stage of progressive hepatic fibrosis and is considered irreversible. In 2010, cirrhosis was the 8th leading cause of death in the U.S., and it most commonly due to Hepatitis C, followed by alcohol.1,2 Acute liver failure can also be due to drugs (most commonly acetaminophen), viral hepatitis, autoimmune, ischemia (shock liver), Wilson’s disease, and idiopathic (up to 14% of causes). Due to the many complications of cirrhosis, patient life expectancy is drastically reduced.1-3
Major complications of cirrhosis include variceal hemorrhage, ascites, spontaneous bacterial peritonitis (SBP), hepatic encephalopathy, hepatorenal syndrome, and hepatopulmonary syndrome. If one of these develops, the patient is considered to have decompensated cirrhosis. Bleeding, infection, increased alcohol intake, dehydration, electrolyte abnormalities (hypokalemia specifically), and constipation can all ultimately contribute to the development of decompensated cirrhosis.2,3
Pathologic changes in the liver lead to the above complications. These pathologic changes include fibrosis and the formation of regenerative nodules that replace normal hepatic tissue. This decreases hepatic venous flow and elevates portal pressures. This in turn causes splenomegaly, resulting in anemia, hypoalbuminemia, thrombocytopenia, and often ascites.1-4
Approach to Initial Resuscitation: Once you walk into the room of the sick cirrhotic patient, always start with the ABCs. If you are alone, you will need to systematically go through the ABCs yourself. If you have a team of EMTs, techs, and nurses, then you can lead the resuscitation and the ABCs can be obtained simultaneously.
- Ensure that the patient is responsive. If the patient is unresponsive and has no pulse, begin Advanced Cardiac Life Support (ACLS) measures.
- Ensure that the patient is protecting his or her airway. If the patient has massive hematemesis or shows poor mentation, have a low threshold to initiate advanced airway measures (see below).
- Check the lungs and breath sounds for equal chest rise and to evaluate for other abnormal breath sounds.
- Intravenous access is critically important in these patients, and ensuring bilateral large bore IV access is essential. If you cannot obtain IV access, then acquire an intraosseous (IO) line. In the initial resuscitation stages, IV and IO access are better than central access.
- For circulation, repeat VS and BP readings liberally. If the patient is hypotensive and not bleeding, an IV fluid bolus may help. Adequate perfusion should be measured by clinical factors such as mental status, capillary refill, pulse pressure, HR, pulse strength, urine output and mean arterial pressure (MAP). Using more than one clinical factor is best. If the patient is bleeding, start a transfusion with 1:1:1 ratio of pRBC:FFP:platelets. These patients often are coagulopathic. Replacing fibrinogen (if less than 100mg/dL) with cryoprecipitate can also be helpful. Tranexamic acid (TXA) may also be useful.
- Ensure you adequately expose the patient and look for signs of infection.
- Complete a quick neurologic exam (gross cranial nerve, motor, sensory, and cerebellar exams).
- Always check an initial blood glucose, as these patients are often hypoglycemic.
Laboratory work should be drawn. A type and cross should be obtained immediately, as these patients may require a significant amount of blood products. A complete blood count (CBC), lactate, venous blood gas (VBG), liver function tests (LFTs), basic metabolic panel (BMP) ammonia level, coagulation panel, fibrinogen level, and electrocardiogram (ECG) (ischemia can occur with shock and bleeding in these patients) should also be ordered.
With bleeding or any instability, administer ceftriaxone 1 gram (g) IV or cefotaxime 2g IV. If bleeding, an IV octreotide bolus of at 50mcg followed by 50mcg/hour can decrease transfusion needs. If outside of the U.S., one should administer terlipressin 2mg IV instead of octreotide. Please see a prior post on the approach to the unstable gastrointestinal (GI) bleeder by emDocs.net for further information: http://www.emdocs.net/unstable-patient-gi-bleed/
Airway
Intubating these patients can be difficult. The concepts of no desaturation (NO DESAT) and delayed sequence intubation (DSI) can be beneficial in decreasing morbidity and mortality. Pre-oxygenating and de-nitrogenating the lungs prior to intubation will provide an oxygen reservoir prior to intubation. This is particularly helpful in patients like the cirrhotic patient, who may desaturate more quickly during intubation. To perform DSI, place the patient on supplemental oxygen by NC. If needed, add a facemask with 15 liters (L) O2. If the patient continues to demonstrate low oxygen levels, non-invasive positive pressure ventilation (NIPPV) may be considered. However, use caution and avoid NIPPV in patients with active hematemesis. Ketamine is a useful medication to administer during DSI as it helps reduce patient discomfort and agitation, allowing for adequate pre-oxygenation and intubation. Using ketamine will also allow you to place a nasogastric tube, which will decrease aspiration risk and clear the stomach of any present blood. A NG tube should only be used to clear the stomach of potential aspiration material and not to diagnose an upper gastrointestinal (GI) bleed.
EMCrit (www.emcrit.org) provides an excellent summary on intubating patients with GI bleeding. This can work with any sick cirrhotic patient. The steps include:
(1) Empty the stomach using a NG tube, and administer metoclopramide 10mg IV.
(2) Intubate the patient with the head of the bed elevated to 45 degrees. Have suction ready.
(3) Ensure adequate pre-oxygenation. Without pre-oxygenation, these patients may rapidly desaturate once medications are provided.
(4) Use smaller doses of the induction and sedation medications. Many of these patients will already be hypotensive and/or altered and thus, will require lower doses of sedation medication (s).
(5) On the other hand, use higher doses of the paralytic medication given during induction and intubation. Paralytic medications such as rocuronium will also augment lower esophageal tone.
(6) Maximize your equipment and have backup techniques and tools ready. These include a videoscope, direct laryngoscope, bougie, laryngeal mask airway (LMA), suction set up, and a meconium aspirator.
(7) If you fail the first attempt, bag slowly and gently, and consider placing an LMA.
(8) If the patient vomits, place him or her in Trendelenburg position to keep emesis contents out of the lungs.
(9) The meconium aspirator can be attached to the endotracheal tube (ETT) for improved suction if your baseline suction device is weaker.
(10) Expect the patient to aspirate with the intubation and be prepared for massive systemic inflammatory response (SIRS).5
History and Exam
If possible, assess the patient’s full medical history including his or her normal weight, chronic medications, follow up visits, prior complications of cirrhosis and endoscopic records. Other important questions to ask include any anorexia, fever, abdominal pain, pruritis, melena, and hematemesis.
After a primary survey and full vital signs have been assessed, a full exam should be conducted. Look for any signs of cerebral edema and herniation (Cushing’s reflex and/or posturing). Volume status can be difficult to ascertain in these patients due to abnormal volume distribution. Test for asterixis (raising both arms with wrists hyperextended) and jaundice (best found under the tongue and sclera). Ascites can be difficult to diagnosis based on exam alone, but ultrasound can be helpful in this setting. Signs of chronic liver failure include ascites, caput medusa (superficial periumbilical veins), muscular atrophy, gynecomastia, testicular atrophy, spider angiomas, palmar erythema, and parotid gland enlargement.3,6
Ascites
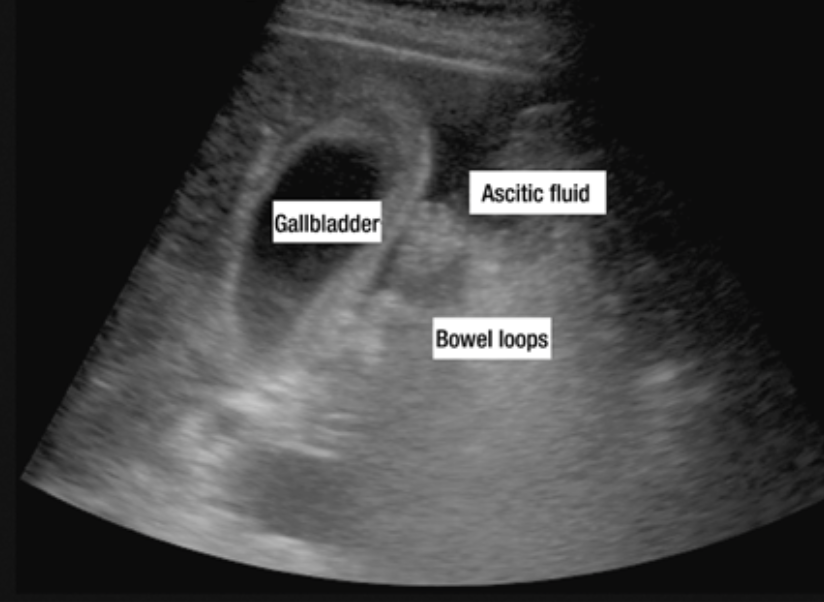
Ascites, or the accumulation of fluid in the abdomen, is the most common complication of cirrhosis. Close to 60% of cirrhotic patients will have ascites within 10 years of diagnosis. Portal hypertension is the first step in fluid accumulation.6,7 When a patient presents with concerning signs for ascites, one should question him or her regarding weight gain and abdominal girth. These both have positive likelihood ratios (LR) of 3.2 and 4.16, respectively.8 A Shifting dullness on exam also has a positive LR of 5.76, while a fluid wave has the highest LR of 9.6.8 No ankle swelling has a negative LR of 0.10 and no change in abdominal girth has a LR of 0.17. However, 1500 mL of ascitic fluid is required for several of these findings. Ultrasound, as demonstrated below, can also assist in diagnosing ascites.8
Chronic management of ascites includes diuresis and sodium restriction. Usually, spironolactone and furosemide are prescribed in a 100:40 ratio. Abstaining from alcohol should be encouraged, as this will drastically improve ascites. These patients ultimately require GI/primary care for medical management.6,7,9
Indications for paracentesis in the ED setting include new-onset ascites, suspected spontaneous bacterial peritonitis (SBP) and/or relief of cardiorespiratory or GI manifestations of tense ascites. Specifically, removal of more than 5 L can assist in improving dyspnea and early satiety. Paracentesis may also be associated with collateral advantages, such as a reduction in hepatic venous pressure gradients, intravariceal pressure, and variceal wall tension, which can in turn decrease risk of GI bleeding. It is important to note that there is no level of international normalized ratio (INR) or platelets that serves as a contraindication for paracentesis.10
GI Bleeding/Hematemesis
Patients with GI bleeding in the setting of ascites typically present with hematemesis and/or melena due to variceal hemorrhage, gastritis, or ulcer. Varices form at a rate of 5-15% per year in cirrhotic patients, with 1/3 of patients eventually suffering hemorrhage. Variceal hemorrhage has an initial mortality rate of >20%, but can be eventually as high as 70%. 6-8 In cirrhotic patients, varices are the cause of bleeding in 50-90% of patients. As described above, airway protection, control of bleeding and avoidance of complications are vital. Only 50% of patients with variceal bleeds will stop bleeding spontaneously, so it is important to manage bleeding vigilantly.11-14
Three management goals of the bleeding cirrhotic patient exist: (1) hemodynamic resuscitation, (2) bleeding cessation and (3) prevention/treatment of complications. Resuscitation including IV access and blood product transfusion is key in initial management, but beware of over-transfusion and volume overload. These patients often demonstrate low blood pressures at baseline, so do not overcorrect, as this can actually worsen bleeding. Goals of transfusion if active exsanguination is present include a hemoglobin (Hgb) of at least 7g/dL and platelets of 50,000/mm3. Have low threshold to transfuse to reach these goals with fresh frozen plasma in a ratio of 1:1:1.11-14 A recent article by Villanueva et al recommends a target hemoglobin goal of 7.0.15 It should, however, be mentioned that this study did not include patients with active, severe hemorrhage.15
Complications of cirrhosis most commonly include infection but also may include aspiration, hematemesis, encephalopathy and renal failure. The administration of antibiotics will dramatically decrease mortality and even recurrent bleeding risk. Common infections include urinary tract infections (UTI) (29%), SBP (23%), and respiratory infections (11%). Administering antibiotics such as ceftriaxone 1g or cefotaxime 2g is essential, as up to 20% of patients with cirrhosis will have an infection at time of admission. In addition, up to half of patients with cirrhosis will develop infection over the course of hospitalization. These patients are also at high risk for aspiration. Although it is controversial as to whether endotracheal intubation increases risk of aspiration, it should be completed to protect the airway in the setting of severe hematemesis. Placement of a NG tube can assist in decompressing the stomach and clearing blood. These patients are also at increased risk of encephalopathy and renal failure with variceal bleeding (these will be covered later). 11,16-18
Control of bleeding is essential. Ultimately, endoscopic therapy should be conducted as soon as possible, as the source of bleeding must be targeted. Esophagogastroduodenoscopy (EGD) should be completed within 12 hours of the development of bleeding. Variceal ligation or sclerotherapy are definitive treatments.11,18,19 This warrants immediate GI, general surgery, and interventional radiology (IR) consultation. All three should be consulted for any severely ill and bleeding cirrhotic patient. Erythromycin can assist gastric emptying and improve EGD success. Vasoactive agents such as octreotide, vasopressin, somatostatin, and terlipressin have also been shown to improve hemostasis, transfusion requirements, and duration of hospitalization. Octreotide at 50mcg bolus with 50mcg/hr drip can decrease portal pressures and azygos blood flow, while also increasing m mean arterial pressure (MAP). Vasopressin at 0.2 to 0.4 units/minute IV with nitroglycerin at 40 micrograms/min IV is another regimen. While not available in the U.S., terlipressin (2mg IV bolus every four hours), is the only actual agent beyond antibiotics that decreases mortality.18-24
If unable to control bleeding with initial measures (endoscopic therapies fail 10% of the time), balloon tamponade is an option. However, this procedure is associated with serious complications such as esophageal rupture and thus, the risks and benefits must be weighed. Three balloons are available including the Sengstaken-Blakemore tube (with a 250cc gastric balloon, esophageal balloon, and single gastric suction port), the Minnesota tube (modified Sengstaken-Blakemore tube with an esophageal suction port above the esophageal balloon), and the Linton-Nachlas tube (single 600cc gastric balloon).22,24,25 At this point, IR and general surgery must be consulted. Transjugular intrahepatic portosystemic shunting (TIPS) is also an option in the setting of endoscopic therapy failure.
Please see EMCrit’s video on Blakemore tube placement: http://emcrit.org/procedures/blakemore-tube-placement/
Spontaneous Bacterial Peritonitis
SBP is defined as an infection of ascitic fluid. Most cases are due to E. coli and/or Klebsiella. SBP is usually a sign of end-stage liver disease. Classic signs of this disease include fever (most common), abdominal pain, and altered mental status. Abdominal pain and altered mental status are often subtle, and because these patients are baseline hypothermic, a definition for fever in these patients should be 37.8oC. On exam, any abdominal tenderness, increase in ascites, or hypotension should raise one’s suspicion for SBP. Other signs and symptoms include diarrhea, ileus, hypothermia, acidosis, and azotemia. If the patient has a temperature greater than 37.8oC, abdominal pain/tenderness, change in mental status, or ascitic neutrophil (PMN) count greater than 250 cells/mm3, then perform a paracentesis and start antibiotics.6,26-30 Each hour in delay of paracentesis increases mortality by 3.3%.28
SBP is diagnosed with an ascitic fluid PMN count ≥250 cells/mm3, > 1000 white blood cells (WBC), positive culture results, and exclusion of any secondary cause (such as a surgical infection). Send the ascitic fluid for cell count/differential, culture, Gram stain, albumin, glucose, protein, LDH, amylase, and bilirubin. Send at least 20 cc of ascitic fluid in two separate blood culture bottles, which can increase yield by 25%.29,32-34
Once cultures are obtained, start antibiotics immediately (either cefotaxime 2g IV or ceftriaxone 1g IV). Cefotaxime 2g IV every 8 hours provides high antibiotic levels in the ascitic fluid. Renal failure develops in up to 40% of patients with SBP, and this can be decreased with administration of IV albumin infusion at 1.5g/kg.30,31
There has been literature on utilizing urine dipstick to quickly diagnose SBP if the dipstick returns positive for leukocyte esterase and/or nitrates. However, sensitivities vary from 31% to 100%, with specificities of 81% to 100%. This may help rule in the diagnosis of SBP, but it cannot be used to rule out disease.31-33A dipstick device designed specifically for diagnosing infection of ascitic fluid is currently in development.34
Other tests of peritoneal fluid have shown promise in diagnosing SBP. An ascites lactate level of greater than 25mg/dL has demonstrated sensitivity and specificity approaching 100%. Along with ascitic fluid neutrophils, a peritoneal fluid pH < 7.35 has also shown promise in diagnosing SBP33,34 Lactoferrin (an iron binding protein present in PMNs) presence in ascitic fluid has been shown in one study to have a sensitivity of 96% and specificity of 87%. However, this test requires further validation.35
Hepatic Encephalopathy
Patients with encephalopathy usually have advanced liver disease with classic exam findings. Encephalopathy is a spectrum of neurologic abnormalities in patients with cirrhosis. It is thought to be due to excess cerebral ammonia elevation, leading to increased glutamine. Overt encephalopathy develops in up to 45% of patients with cirrhosis, but minimal encephalopathy with subtle findings will be present in 80% of patients.36-39 Grading encephalopathy is based on four levels as depicted below in a table derived from MDcalc:

One of the earliest markers of hepatic encephalopathy is a disturbance in sleep patterns, specifically hypersomnia during the day and insomnia at night. Other signs include asterixis, hyperactive reflexes, and decerebrate posturing. Asterixis may be present in Grade 1, but it is often pronounced at Grade II. Asterixis is defined as flapping motion of outstretched, dorsiflexed hands.36-39 Focal neurologic deficits (most commonly hemiplegia) can also be present in up to 17%, despite normal imaging.41 Overt encephalopathy, if present with severe cerebral edema, can lead to coma.36,37,41
Diagnosis of hepatic encephalopathy requires the following: history and exam supportive of the disease, exclusion of other causes of mental status changes (labs and head CT are recommended), and search for precipitating causes. In terms of history, ask caregiver(s), if possible, if there are any new changes in sleep patterns and how the patient’s current condition is different than his or her baseline.36,,37,4 An elevated ammonia level is not required for the diagnosis, though it often is elevated. Ammonia is elevated in liver disease due to decreased liver function and clearance from the blood, but many other causes of ammonia elevation can be present.36,40-43 In general, ammonia levels do not correlate with degree of encephalopathy.44
Precipitants of encephalopathy include GI bleeding, infection (SBP, UTI), hypokalemia, renal failure, dehydration, hypoxia, sedatives, hypoglycemia, and constipation. Evaluation should target these causes.36,37,41
Labs include CBC, LFTs, RFP, ammonia, VBG, ECG, UA, and bilirubin. A non-contrast head CT should be strongly considered, and it may reveal generalized or localized cerebral edema.
Treatment of hepatic encephalopathy first focuses on reversing predisposing conditions such as infection (SBP, UTI), GI bleeding, electrolyte abnormalities (hypokalemia in particular), and constipation. After finding and correcting the precipitant, decreasing cerebral and blood ammonia concentrations is next. Lactulose at 30 to 45ml 2-4 times per day by mouth is most efficacious. If a patient is not PO tolerant, a lactulose enema can be used (1-3L of 20% solution). If a patient is still not improving, rifaximin 400mg three times daily added to lactulose can be beneficial. Neomycin can be used as second line therapy, but it does not have randomized trial support and can cause ototoxicity and nephrotoxicity.41,43-45 Polyethylene glycol does have support for excreting ammonia in the stool with 4L given over 4 hours.45 Other treatments that may work but are currently under study include flumazenil, L-carnitine, acarbose, sodium benzoate, melatonin, serotonin antagonists, and opioid antagonists.41-47
These patients are often agitated and can be a hazard to themselves and others. If a patient becomes very agitated, soft restraints and redirection should be first utilized. Haldol may be given but benzodiazepines should be avoided. The newer generation antipsychotics do not currently have much evidence for use in this population.41,44-46
Most patients with grade I encephalopathy can be discharged with outpatient management, but disposition of patients with grade II encephalopathy depends on the patient’s clinical status. If a patient will not be able to adhere to treatment based on social situation, then admission is prudent. Grades III and IV encephalopathic patients should be admitted, usually to an ICU.41,45
Hepatorenal syndrome
This disease is diagnosed when renal failure is present in the setting of advanced liver disease. This is a diagnosis of exclusion. It will develop in 39% of patients within five years and is a marker of liver failure. It is due to reductions in renal blood flow, caused by splanchnic vasodilation and increased nitric oxide. This decrease in renal perfusion reduces GFR and sodium excretion. Precipitants of hepatorenal synddrome include GI bleeding, infection, dehydration, electrolyte derangements, and renal injury.48,49
Hepatorenal syndrome is marked by little to no urine sediment, low urine sodium, an increase in serum creatinine (Cr), and oliguria. Oliguria is controversial, as some patients will maintain urine output of greater than 400ml/day. As these findings are similar to the diagnosis of acute tubular necrosis and prerenal azotemia, hepatorenal syndrome is also diagnosed with urine red cells less than 50/high power field, protein excretion less than 500mg/day, and no improvement with IV albumin. 48,50-52 Biopsy may be needed.
There are two types of hepatorenal syndrome: Type 1 is the most serious with a Cr above 2.5, or doubling of Cr in a period of less than 2 weeks. Type two is less severe and usually develops over a period of greater than two weeks.48,50-52 Remember, these are diagnoses of exclusion.
For sick, unstable/hypotensive patients, initial treatment with norepinephrine at 0.5-3mg/hr to raise MAP by 10mmHg should be started. Albumin at 1g/kg per day should also be administered and given for a total of two days. If the patient is stable, a combination of midodrine at 7.5mg PO TID, octreotide 50mcg/hr IV, and albumin 1g/kg should be done (this is the recommended treatment for rehydration, though one of the diagnostic criteria is no response to albumin). Transjugular intrahepatic portosystemic shunt (TIPS) is sometimes successful for nonresponders, but ultimately liver transplant is required with hemodialysis as a bridge to transplant.48, 50-52
Hepatopulmonary syndrome
This is defined as liver disease with an increased A-A gradient while on room air, and evidence of intrapulmonary vascular abnormalities (IPVDs). IPVDs are diagnosed with nuclear scanning, contrast echocardiography, and/or pulmonary arteriography. Hepatopulmonary syndrome is present with portal hypertension but does not require cirrhosis.53,54 The IPVDs include increased pulmonary dilation of vessels and increased number of pulmonary vessels. The prevalence of IPVDs ranges from 4-47% of cirrhotic patients, with mortality at 5 years of 75%. Surprisingly, the cause of death is usually due to complications of cirrhosis rather than respiratory failure. Unfortunately, mild hypoxemia is common in ascites due to diaphragm elevation, decreasing FRC and increasing V/Q mismatch. Thus, making the diagnosis can be difficult.53-56
Classic findings of hepatopulmonary syndrome include platypnea (increase in dyspnea when upright, improved with recumbency) and orthodeoxia (decrease in arterial O2 when moving from supine to upright position). These are thought to occur due to increased blood flow to IPVDs.56,57
Liver transplant is the only means of a cure. TIPS does have some support with case reports. There is no medical treatment shown to treat this condition besides optimizing ascites and other medical conditions. Oxygen supplementation can be used to treat baseline hypoxemia and symptoms such as shortness of breath. 53,56-59
Liver Transplant Criteria
For transplantation to be considered, patients with cirrhosis must have a complication of portal hypertension and severe disease. Severe variceal hemorrhage, ascites, encephalopathy, and hepatorenal disease are manifestations of end stage disease and are markers for transplant. Tools including the Model for End-stage Liver Disease (MELD) and Child-Turcotte-Pugh system can be used to evaluate patients for transplant. MELD incorporates age, creatinine, bilirubin, INR, and hemodialysis 2X in the last week. Scores greater than 10 warrant evaluation for transplant, while patients with scores greater than 15 are candidates.60 The Child-Turcotte-Pugh (CTP) system is also used to assess prognosis in chronic failure but was initially developed to assess risk with shunt surgery in variceal bleeding. This contains classes A, B, and C (C has the worst mortality), with points ranging from 1-3 for the following: encephalopathy, ascites, bilirubin, albumin, and PT/INR. Five to six points is Class A, 7-9 Class B, and 10-15 Class C.61
References/Further Reading
- Murray CJ, Atkinson C, Bhalla K, et al. The state of US health, 1990-2010: burden of diseases, injuries, and risk factors. JAMA 2013; 310:591.
- Singal AK, Anand BS: Recent trends in the epidemiology of alcoholic liver disease. Clin Liver Dis 2: 53, 2013.
- Ichai P, Samuel D: Epidemiology of liver failure. Clin Res Hepatol Gastroenterol 35: 610, 2011.
- Patel A. The unstable patient with a gastrointestinal bleed. emDocs.net. 21 Dec 2014. http://www.emdocs.net/unstable-patient-gi-bleed/.
- Weingart S. EMCrit Podcast 5 – Intubating the Critical GI Bleeder. EMCrit. 21 June 2009. http://emcrit.org/podcasts/intubating-gi-bleeds/.
- Ginés P, Quintero E, Arroyo V, et al. Compensated cirrhosis: natural history and prognostic factors. Hepatology 1987; 7:122.
- Runyon BA, AASLD Practice Guidelines Committee. Management of adult patients with ascites due to cirrhosis: an update. Hepatology 2009; 49:2087.
- Simel DL, et al. Quantitating bedside diagnosis: clinical evaluation of ascites. J Gen Intern Med. 1988 Sep-Oct;3(5):423-8.
- Fogel MR, Sawhney VK, Neal EA, et al. Diuresis in the ascitic patient: a randomized controlled trial of three regimens. J Clin Gastroenterol 1981; 3 Suppl 1:73.
- Runyon MS and Marx JA. Peritoneal Procedures. Roberts and Hedges’ Clinical Procedures in Emergency Medicine, 6th edition: Chapter 43; 852-872.e3.
- Smith JL, Graham DY. Variceal hemorrhage: a critical evaluation of survival analysis. Gastroenterology 1982; 82:968.
- Graham DY, Smith JL. The course of patients after variceal hemorrhage. Gastroenterology 1981; 80:800.
- .D’Amico G, De Franchis R, Cooperative Study Group. Upper digestive bleeding in cirrhosis. Post-therapeutic outcome and prognostic indicators. Hepatology 2003; 38:599
- Krige JE, Kotze UK, Distiller G, et al. Predictive factors for rebleeding and death in alcoholic cirrhotic patients with acute variceal bleeding: a multivariate analysis. World J Surg 2009; 33:212
- Villanueva C, Colomo A, Bosch A, et al. Transfusion strategies for acute upper gastrointestinal bleeding. N Engl J Med 2013; 368:11
- Soares-Weiser K, Brezis M, Tur-Kaspa R, Leibovici L. Antibiotic prophylaxis for cirrhotic patients with gastrointestinal bleeding. Cochrane Database Syst Rev 2002; CD002907
- Hou MC, Lin HC, Liu TT, et al. Antibiotic prophylaxis after endoscopic therapy prevents rebleeding in acute variceal hemorrhage: a randomized trial. Hepatology 2004; 39:746
- Habib A, Sanyal AJ. Acute variceal hemorrhage. Gastrointest Endosc Clin N Am 2007; 17:223
- Hwang JH, Shergill AK, Acosta RD, et al. The role of endoscopy in the management of variceal hemorrhage. Gastrointest Endosc 2014; 80:221
- Garcia-Tsao G, Sanyal AJ, Grace ND, et al. Prevention and management of gastroesophageal varices and variceal hemorrhage in cirrhosis. Hepatology 2007; 46:922
- Wells M, Chande N, Adams P, et al. Meta-analysis: vasoactive medications for the management of acute variceal bleeds. Aliment Pharmacol Ther 2012; 35:1267
- D’amico G, Criscuoli V, Fili D, et al. Meta-analysis of trials for variceal bleeding. Hepatology 2002; 36:1023
- Seo YS, Park SY, Kim MY, et al. Lack of difference among terlipressin, somatostatin, and octreotide in the control of acute gastroesophageal variceal hemorrhage. Hepatology 2014; 60:954
- D’Amico G, Pagliaro L, Bosch J. The treatment of portal hypertension: a meta-analytic review. Hepatology 1995; 22:332
- Chojkier M, Conn HO. Esophageal tamponade in the treatment of bleeding varices. A decade progress report. Dig Dis Sci 1980; 25:267
- Runyon BA, AASLD. Introduction to the revised American Association for the Study of Liver Diseases Practice Guideline management of adult patients with ascites due to cirrhosis 2012. Hepatology 2013; 57:1651
- Chinnock B, Hendey GW, Minnigan H, et al. Clinical impression and ascites appearance do not rule out bacterial peritonitis. J Emerg Med 2013; 44:903
- Kim JJ, Tsukamoto MM, Mathur AK, et al. Delayed paracentesis is associated with increased in-hospital mortality in patients with spontaneous bacterial peritonitis. Am J Gastroenterol 2014; 109:1436
- Runyon BA, Canawati HN, Akriviadis EA. Optimization of ascitic fluid culture technique. Gastroenterology 1988; 95:1351.
- Chavez-Tapia NC, Soares-Weiser K, Brezis M, Leibovici L. Antibiotics for spontaneous bacterial peritonitis in cirrhotic patients. Cochrane Database Syst Rev 2009; CD002232
- Follo A, Llovet JM, Navasa M, et al. Renal impairment after spontaneous bacterial peritonitis in cirrhosis: incidence, clinical course, predictive factors and prognosis. Hepatology 1994; 20:1495
- Nousbaum JB, Cadranel JF, Nahon P, et al. Diagnostic accuracy of the Multistix 8 SG reagent strip in diagnosis of spontaneous bacterial peritonitis. Hepatology 2007; 45:1275
- Mendler MH, Agarwal A, Trimzi M, et al. A new highly sensitive point of care screen for spontaneous bacterial peritonitis using the leukocyte esterase method. J Hepatol 2010; 53:477
- Téllez-Ávila FI, Chávez-Tapia NC, Franco-Guzmán AM, Uribe M, Vargas-Vorackova F. Rapid diagnosis of spontaneous bacterial peritonitis using leukocyte esterase reagent strips in emergency department: uri-quick clini-10SG® vs. Multistix 10SG®. Ann Hepatol. 2012 Sep-Oct. 11(5):696-9
- Parsi MA, Saadeh SN, Zein NN, et al. Ascitic fluid lactoferrin for diagnosis of spontaneous bacterial peritonitis. Gastroenterology 2008; 135:803
- Gill RQ, Sterling RK. Acute liver failure. J Clin Gastroenterol 2001; 33:191
- Khungar V, Poordad F. Hepatic encephalopathy. Clin Liver Dis 2012; 16:301
- Romero-Gómez M, Boza F, García-Valdecasas MS, et al. Subclinical hepatic encephalopathy predicts the development of overt hepatic encephalopathy. Am J Gastroenterol 2001; 96:2718
- Bajaj JS, Wade JB, Sanyal AJ. Spectrum of neurocognitive impairment in cirrhosis: Implications for the assessment of hepatic encephalopathy. Hepatology 2009; 50:2014
- Cadranel JF, Lebiez E, Di Martino V, et al. Focal neurological signs in hepatic encephalopathy in cirrhotic patients: an underestimated entity? Am J Gastroenterol 2001; 96:515
- Vilstrup H, Amodio P, Bajaj J, et al. Hepatic encephalopathy in chronic liver disease: 2014 Practice Guideline by the American Association for the Study of Liver Diseases and the European Association for the Study of the Liver. Hepatology 2014; 60:715
- Ong JP, Aggarwal A, Krieger D, et al. Correlation between ammonia levels and the severity of hepatic encephalopathy. Am J Med 2003; 114:188
- Nicolao F, Efrati C, Masini A, et al. Role of determination of partial pressure of ammonia in cirrhotic patients with and without hepatic encephalopathy. J Hepatol 2003; 38:441
- Mendenhall C. Alcoholic hepatitis. Clin Gastroenterol. 1981;10:417-441
- Bajaj JS. Review article: the modern management of hepatic encephalopathy. Aliment Pharmacol Ther 2010; 31:537
- Bass NM, Mullen KD, Sanyal A, et al. Rifaximin treatment in hepatic encephalopathy. N Engl J Med 2010; 362:1071
- Rahimi RS, Singal AG, Cuthbert JA, Rockey DC. Lactulose vs polyethylene glycol 3350–electrolyte solution for treatment of overt hepatic encephalopathy: the HELP randomized clinical trial. JAMA Intern Med 2014; 174:1727
- Ginès P, Guevara M, Arroyo V, Rodés J. Hepatorenal syndrome. Lancet 2003; 362:1819
- Wadei HM, Mai ML, Ahsan N, Gonwa TA. Hepatorenal syndrome: pathophysiology and management. Clin J Am Soc Nephrol 2006; 1:1066
- Arroyo V, Ginès P, Gerbes AL, et al. Definition and diagnostic criteria of refractory ascites and hepatorenal syndrome in cirrhosis. International Ascites Club. Hepatology 1996; 23:164
- Velez JC, Nietert PJ. Therapeutic response to vasoconstrictors in hepatorenal syndrome parallels increase in mean arterial pressure: a pooled analysis of clinical trials. Am J Kidney Dis 2011; 58:928
- Arroyo V, Guevara M, Ginès P. Hepatorenal syndrome in cirrhosis: pathogenesis and treatment. Gastroenterology 2002; 122:1658
- Hoeper MM, Krowka MJ, Strassburg CP. Portopulmonary hypertension and hepatopulmonary syndrome. Lancet 2004; 363:1461
- Krowka MJ, Cortese DA. Hepatopulmonary syndrome. Current concepts in diagnostic and therapeutic considerations. Chest 1994; 105:1528
- Schenk P, Schöniger-Hekele M, Fuhrmann V, et al. Prognostic significance of the hepatopulmonary syndrome in patients with cirrhosis. Gastroenterology 2003; 125:1042
- Seward JB, Hayes DL, Smith HC, et al. Platypnea-orthodeoxia: clinical profile, diagnostic workup, management, and report of seven cases. Mayo Clin Proc 1984; 59:221
- Berthelot P, Walker JG, Sherlock S, Reid L. Arterial changes in the lungs in cirrhosis of the liver–lung spider nevi. N Engl J Med 1966; 274:291
- Benítez C, Arrese M, Jorquera J, et al. Successful treatment of severe hepatopulmonary syndrome with a sequential use of TIPS placement and liver transplantation. Ann Hepatol 2009; 8:71
- Goldberg DS, Krok K, Batra S, et al. Impact of the hepatopulmonary syndrome MELD exception policy on outcomes of patients after liver transplantation: an analysis of the UNOS database. Gastroenterology 2014; 146:1256
- Martin P, DiMartini A, Feng S, et al. Evaluation for liver transplantation in adults: 2013 practice guideline by the American Association for the Study of Liver Diseases and the American Society of Transplantation. Hepatology 2014; 59:1144
- Child CG, Turcotte JG. Surgery and portal hypertension. In: The liver and portal hypertension. Edited by CG Child. Philadelphia: Saunders 1964:50-72.





4 thoughts on “Approach to the Sick Cirrhotic Patient”
Pingback: emDOCs.net – Emergency Medicine EducationThe EM Educator Series: When sepsis care becomes not so straightforward - emDOCs.net - Emergency Medicine Education
Pingback: emDOCs.net – Emergency Medicine EducationEM@3AM: Gastroesophageal Varices - emDOCs.net - Emergency Medicine Education
Pingback: Management of the Massive GI Bleed - First10EM
Pingback: October Asynchronous Learning – Lakeland Health EM Blog