
Initial Hematology Work Up for Cytopenias
How should you work up anemia, leukopenia, or thrombocytopenia?
Initial Hematology Work Up for Cytopenias Read More »
How should you work up anemia, leukopenia, or thrombocytopenia?
Initial Hematology Work Up for Cytopenias Read More »
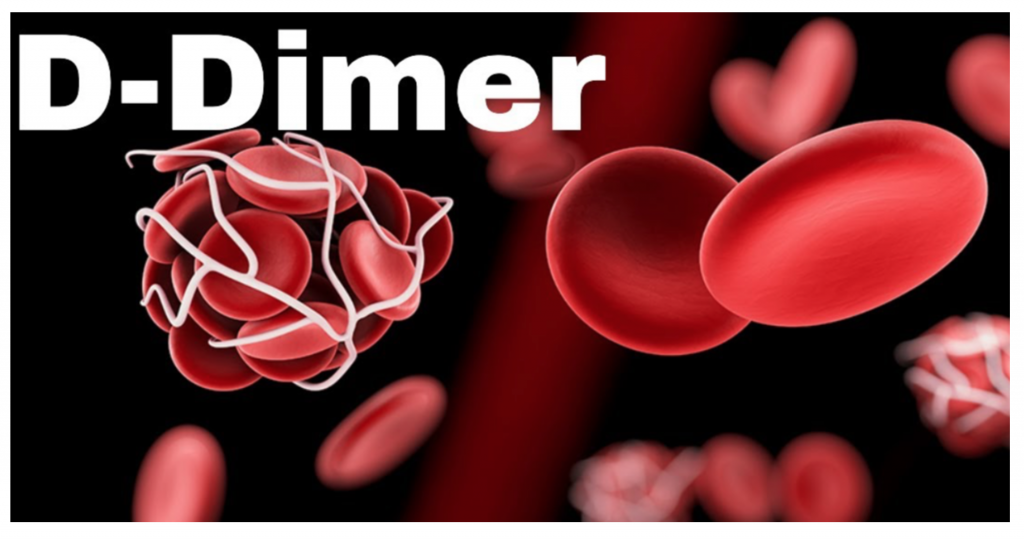
What is a D-dimer, and how should you use this in the ED?
Utility of D-dimer Testing in Special Populations Read More »
The emDOCs cast covers hemophilia this week. What should you consider when it comes to evaluating and managing these patients?
emDOCs Podcast – Episode 81: Hemophilia Read More »
A 3-year-old male with G6PD deficiency presents to your ED by ambulance with shortness of breath, found to be hypoxic with SpO2 80% on 15L NRBM. He appears pale and dyspneic. The child is placed on HFNC with no improvement of saturation. Given his persistent hypoxia on noninvasive methods, a decision is made to intubate. Bedside CXR is unremarkable, and in spite of full ventilatory support with FiO2 of 100%, the patient’s saturation remains at 85%. You later learn he had eaten a full box of blueberries earlier that day. What is the likely diagnosis?
EM@3AM: Acute Hemolytic Anemia Read More »
A 23-year-old female with no past history presents with prolonged bleeding from her tooth extraction earlier the same day. Her dentist was planning on removing all her wisdom teeth but stopped after the first extraction due to inability to achieve hemostasis. She has never experienced this kind of bleeding before but notes that recently her gums often bleed when brushing her teeth and describes her last few menstrual cycles as “heavier” than usual. She is not on any blood thinners and was adopted at birth without record of family medical history. She is stable. Tooth number 17 appears to have been extracted, and there is blood-soaked cotton balls and gauze between the buccal mucosa and the cavity where tooth 17 used to be. Upon removal of the gauze, you notice a slow oozing of blood from the extraction site. What are some of the bleeding disorders on your differential given this clinical presentation?
EM@3AM: Bleeding Disorders Read More »
The emDOCs Podcast covers the challenging diagnosis of cerebral venous thrombosis.
emDOCs Podcast – Episode 37: Cerebral Venous Thrombosis Read More »
A 4-year-old vaccinated male is brought to the ED by his parents for 3 days of hematuria and abdominal pain. They present now because he refuses to ambulate. He has a rash on his legs that family attributes to playing outside in the grass. His vital signs include HR 135, RR 20, and temperature of 37.9 C. His abdomen is diffusely tender, and he has red, raised papules on the buttocks and lower legs. What is the diagnosis, and what is the most common gastrointestinal complication?
A 63-year-old male without any past medical history presents to the emergency department with several weeks of headaches, some blurry vision while walking at a quick pace, and shortness of breath. His last checkup with his primary doctor was about 3 years ago and everything was ‘normal’. He takes no medications, has no allergies, and has no previous surgeries. He admits to being a long-term smoker, about a half a pack a day for 20 years. Examination in the ED is normal. Basic lab work is drawn which reveals a hematocrit of 63%, elevated RBC mass, and thrombocytosis to 550K/ml. On further evaluation as an inpatient, it was found the patient had a low erythropoietin level. What is the diagnosis?
EM@3AM: Polycythemia Read More »

Cerebral venous thrombosis is a tough diagnosis… Why is it missed, and how can we improve diagnosis?
Cerebral venous thrombosis: why we miss it and how we can improve Read More »

What is the literature behind fixed-dose PCC for reversal of vitamin K antagonist-associated bleeding? Can you use this for your next shift?