Today on the emDOCs cast, Brit Long, MD (@long_brit) covers hemophilia, including background, severity, evaluation, and management.
Episode 81: Hemophilia
What is hemophilia?
- Bleeding disorder due to a deficiency in one or more of the proteins (factors) involved in the coagulation pathway.
- Interruption of the coagulation cascade → ↓ fibrin clot formation →
- Several types:
- Hemophilia A – deficiency in factor VIII, 80-85% of cases
- Hemophilia B – deficiency in factor IX
- Hemophilia C – deficiency in factor XI
- Females can be affected.
- Classically thought of as x-linked and only found in males.
- Females who carry the gene can have low factor levels and are at risk of bleeding complications.
- Most have symptoms of mild hemophilia, but they can have severe hemophilia symptoms that require factor replacement.
- Spontaneous mutations occur.
- ~ 1/3 of cases have a spontaneous mutation with no family history.
- Acquired forms are rare.
- Caused by autoantibodies/inhibitors to clotting factors.
- May be associated with other autoimmune conditions, cancer, dermatological disorders, pregnancy, drugs. ~50% are idiopathic.
Severity of hemophilia is based on factor level:
- Severe hemophilia: <1 % factor available
- Moderate hemophilia: factor level 1-5%
- Mild hemophilia: factor level 5-40%
Diagnosed hemophilia presentations:
- Most patients will have a known diagnosis, and the patient/family are usually extremely knowledgeable about the disease.
- Trivial bleeds will typically be managed at home.
- Many patients will bring in their own factor replacement. Use their factor replacement if possible.
Undiagnosed hemophilia presentations:
- Age of the 1st bleeding episode relates to severity of factor deficiency.
- Severe hemophilia – presents birth to 4 years
- In one study, 20% <2 years old presented with intra- or extracranial hemorrhage. Other presenting bleeds were post-circumcision (27%) and heel-stick bleeds (17%).
- A 2013 study found oral mucosal and head bleeding due to injury were most common in those < 6 months. In patients >6 months, hemarthrosis was most common. Once they started walking it was bruising, MSK, and mouth bleeds.
- Mild hemophilia can be undiagnosed for years.
- One study found age of diagnosis ranged from 14 to 62 years.
- Severe hemophilia – presents birth to 4 years
- Pediatric patients:
- Post circumcision bleed, head bleeds, bruises.
- Toddlers may present with joint bleeds, excessive or prolonged bleeding after small cuts and lacerations, and bruises that are larger than what you would expect for the mechanism.
- Adults:
- Spontaneous bruising, ecchymosis, mucosal bleeding, muscle hematomas, subacute or delayed postpartum bleeding, or any other unexplained bleeding.
- Any patient with isolated ↑aPTT but normal PT and platelets.
Bleeding can be spontaneous or associated with trauma
- Presentation depends on the organ system.
- Most common site is hemarthrosis (70-80% of cases).
- Intramuscular bleeding (10-20%)
- CNS bleeding (< 5%)
- Severe bleeding:
- Intracranial hemorrhage (leading cause of death)
- Hematomas involving a major structure (e.g. airway, retroperitoneal, etc)
- May present with bruising, mucosal bleeding, delayed postpartum bleeding, persistent oozing after trauma.
Approach to patient with hemophilia:
- Does the patient have a major or minor bleed?
- Major: CNS, retroperitoneum, throat, neck, eyes, chest, GI tract, hemarthrosis if neurovascular compromise or rapid expansion
- Minor: Joints, muscles, or deep lacerations.
- Treatment depends on the HISTORY.
- Key questions for patient with known hemophilia:
- Type of hemophilia
- Baseline factor level/severity
- If on prophylaxis, which one?
- When was the last factor replacement?
- Any prior history of severe bleeding or spontaneous bleeding?
- Do they have inhibitors or have they been treated for them?
- HIV, hep C status
- Do they have an emergency plan from their hematologist?
- Did they bring factor from home with them?
- Questions for suspected by undiagnosed hemophilia:
- History of prior bleeds disproportionate to injury?
- History of spontaneous atraumatic hemorrhage, ecchymosis, or mucosal bleeding?
- History of intracerebral hemorrhage or hemarthrosis?
- History of familial bleeding disorders?
- Key questions for patient with known hemophilia:
- Exam needs to be thorough, assessing for sites of hematoma.
- Exam may be normal in the early part of the bleeding episode.
- Consider bleeding affecting the CNS, HEENT, MSK system, GI, RP, chest. Look for bruising, swelling, and mucosal bleeding.
- Head bleeds can be spontaneous with no history of trauma. If concerning mechanism even with a normal examination, give factor concentrate and then get imaging with CT.
- Retroperitoneal (RP) hematomas can present with vague back pain, flank pain, abdominal pain, or groin pain. Pain often worse with external rotation of the hip.
- For deep muscle hematomas and RP bleed, fully expose the patient to look at the abdomen, flank, and all the muscle compartments.

Lab testing
- If known hemophilia diagnosis:
- Labs play no significant role in the acute management and should never delay indicated coagulation correction.
- If the patient does not know their factor level, assume a level of zero.
- Obtain a CBC, coag profile, and factor activity level if possible to help hematology assess necessary factor dosing changes.
- If suspected hemophilia:
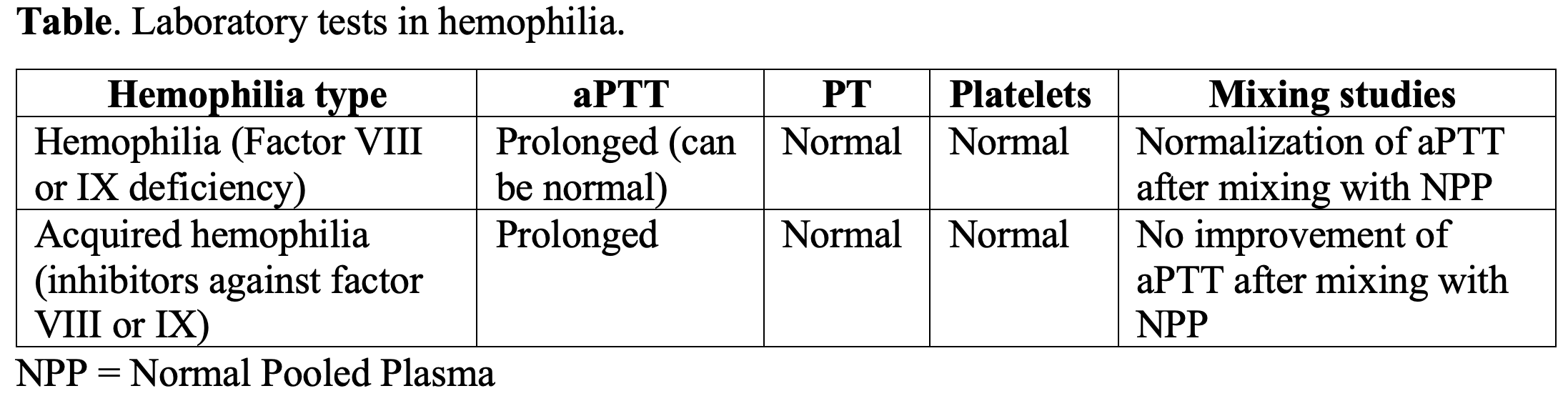
- May see prolonged aPTT, with a normal PT, bleeding time, and platelet count.
- However, a normal aPTT does not rule out hemophilia.
- A mixing study may help determine if an inhibitor is present. The patient’s plasma is mixed with normal pooled plasma (NPP) which adds sufficient clotting factors to overcome the deficiency. If an inhibitor is present, it will inhibit the clotting factors in patient plasma and the NPP, and the clotting time remains prolonged.
Decision rules and hemophilia
- Have a low threshold for imaging based on the history and exam.
- Clinical decision tools (e.g PECARN Pediatric Head Injury/Trauma Algorithm, Canadian CT Head Injury Rule, Ottawa Knee and Ankle Rules) do not apply to those with hemophilia and should not guide management.
- Do not delay factor replacement for imaging.
Management:
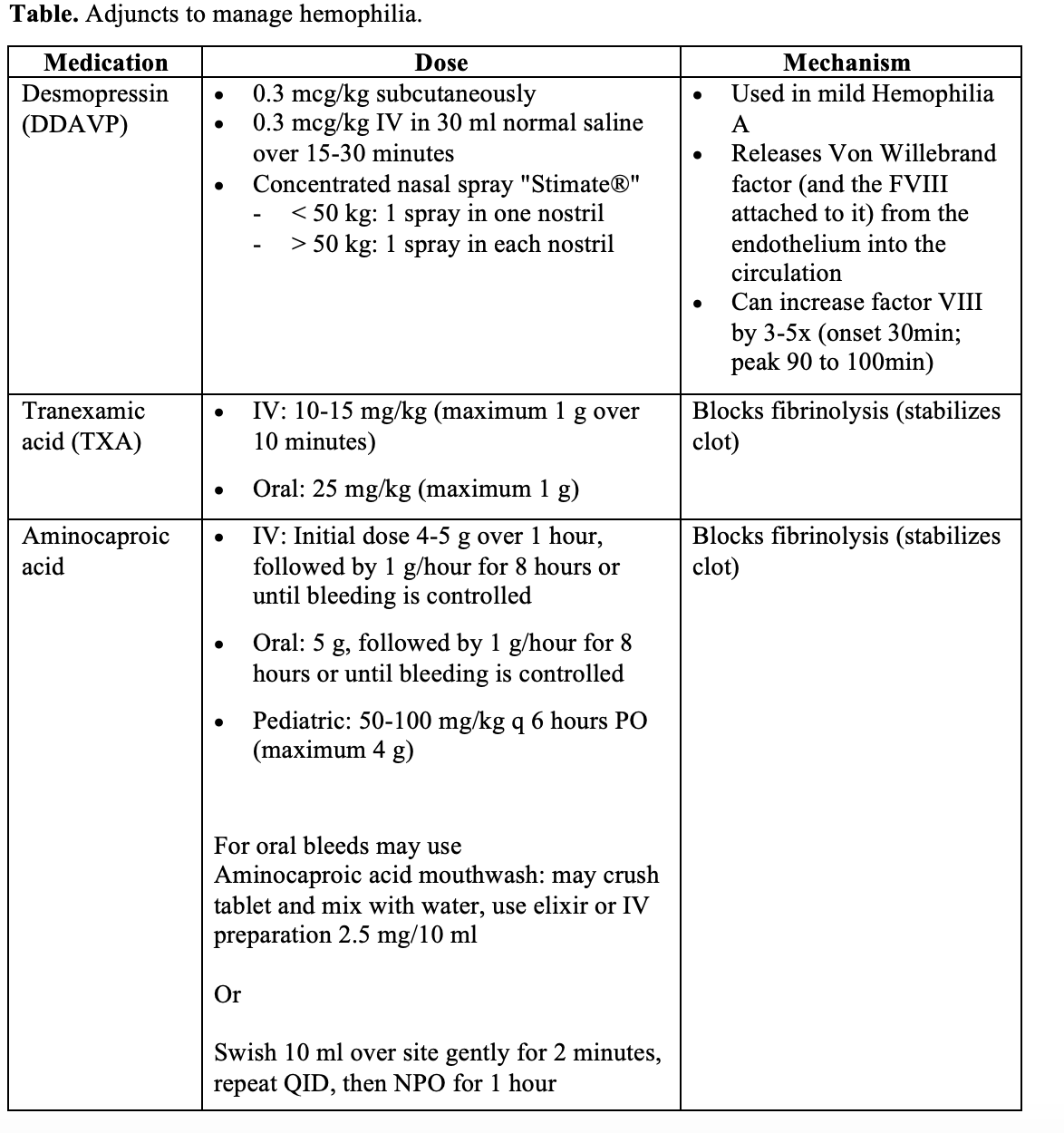
- Control bleeding
- If superficial bleeding, apply localized pressure and topical thrombin products.
- For a deep laceration or continued bleeding despite above measures, use desmopressin, tranexamic acid, and aminocaproic acid.
- Replace coagulation factors
- For major bleeding, raise the clotting factors rapidly. Do not wait for labs or images. Goal is a factor level of 100% for major bleeds and up to 50% for minor bleeds.
Indications for factor replacement therapy (Source here).

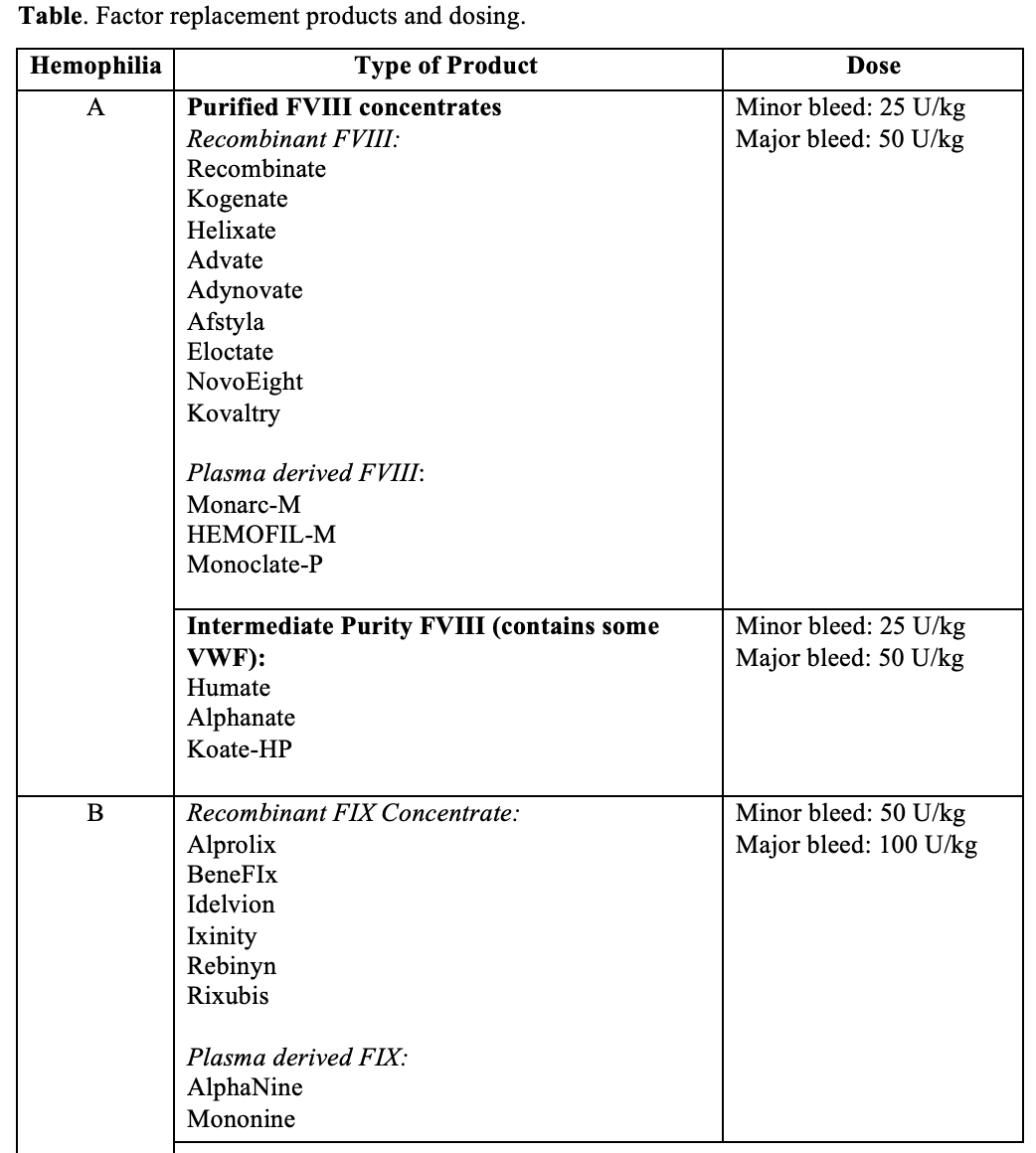
Choosing the right factor concentrate and dosing:
- There are several factor concentrates available, and they differ in their purification and derivation.
- Dosing for all of them is the same.
- Each unit of FVIII/kg raises the plasma FVIII level by 2%. To raise the circulating FVIII level to 100%, the replacement dose is 50 U/kg.
- Each unit of FIX/kg raises the plasma FIX by 1%. To raise the circulating level to 100% give a replacement dose of 100 U/kg.
- If you’re concerned about bleeding and you don’t know the patient’s baseline factor level assume the level is 0%. Excess factor concentrate will not cause a hypercoagulable state.
Is lumbar puncture safe in patients with hemophilia?
- If LP is absolutely necessary, pretreat with clotting factors and speak with hematology.
- Other procedures needing factor replacement: arterial line placement, intramuscular injection, and arthrocentesis.
- Fractures and dislocations are indications for full dose factor replacement. This includes something as minor as a nursemaid’s elbow.
What medications should be avoided?
- Avoid medications that affect the clotting cascade or formation of the platelet plug: aspirin, Alka-Seltzer, Pepto-Bismol, many cough medicines, NSAIDs, anticoagulants and antiplatelet medications.
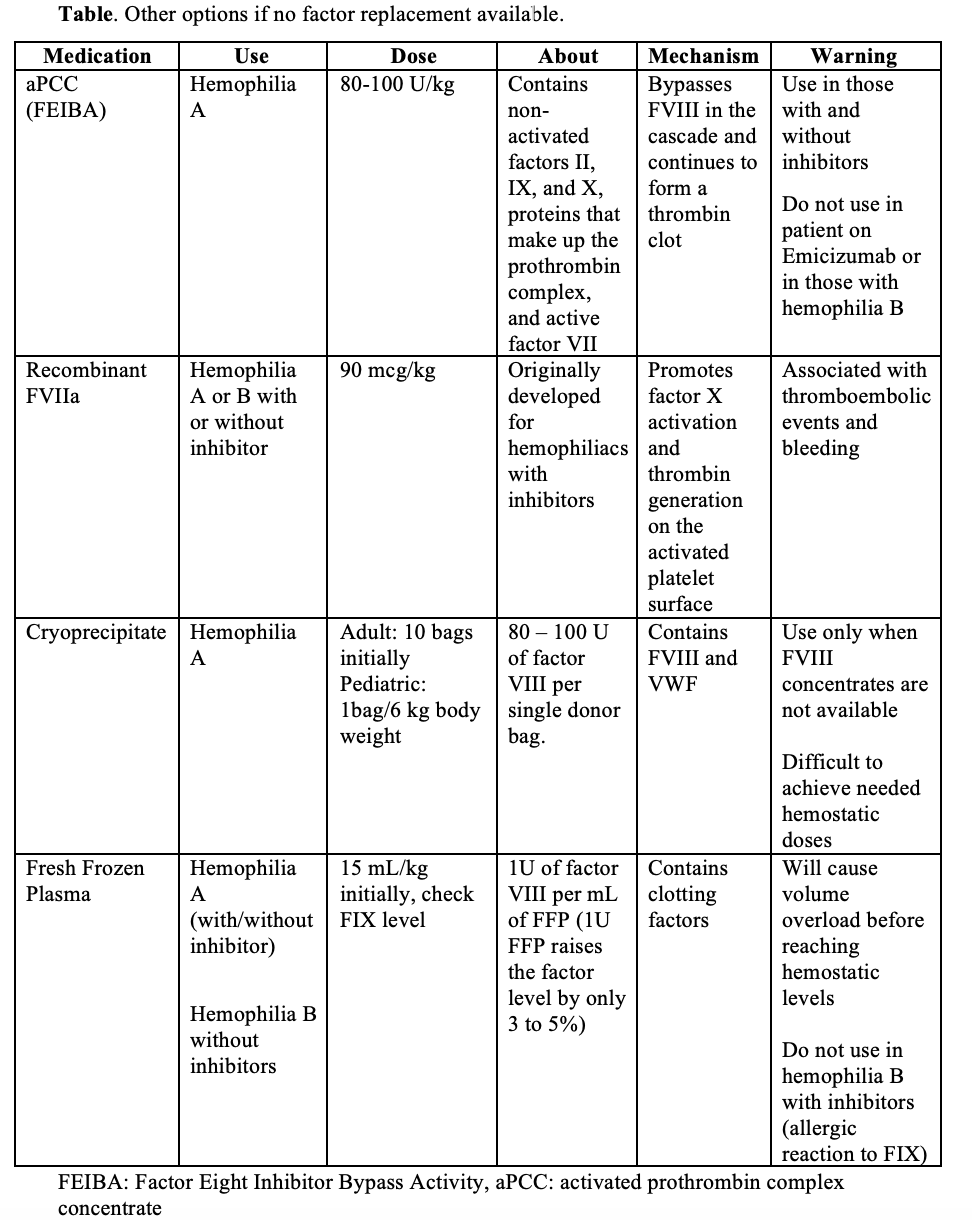
What should we consider in those with inhibitors?
- One of the more serious complications of hemophilia treatment is the development of anti-factor antibodies that inhibit FVIII or FIX. This occurs in 1 in 5 people with hemophilia A and 3 in 100 people with hemophilia B.
- Inhibitors develop against the treatment product, most often within the first 50 days of starting factor replacement. Inhibitors interfere with the patient’s own circulating factors which makes the disease more severe, and they interfere with infused factor concentrates.
- Diagnosing an inhibitor is challenging. Consider it if the patient has a history of recurrent or breakthrough bleeds. Diagnosis focuses on measuring the titer of antibodies in the patient’s blood (titer >5 is high).
- Treatment is also challenging. For life or limb threatening bleeding and known inhibitor, the safest treatment is recombinant factor VIIa (90 mcg/kg) for hemophilia A or B. Activated prothrombin complex concentrate is an option for hemophilia A, unless they are on emicizumab.

Pearls:
- Hemophilia is a bleeding disorder due to a deficiency in coagulation factor VIII (in hemophilia A) or IX (in hemophilia B).
- The mainstay of managing patients is the immediate replacement of clotting factors based on the history and suspicion of bleeding and not exam, labs, or imaging.
- We need to have a low threshold for imaging based on the underlying history.
- The doses for factor concentrate replacement to achieve 100% factor levels are 50 U/kg for factor VIII and 100 U/kg for factor IX.
References:
- Alblaihed L, et al. High risk and low prevalence diseases: Hemophilia emergencies. Am J Emerg Med. 2022;56:21-27. PMID: 35349958.
- Kulkarni R, et al; Haemophilia Treatment Center Network Investigators. Sites of initial bleeding episodes, mode of delivery and age of diagnosis in babies with haemophilia diagnosed before the age of 2 years: a report from The Centers for Disease Control and Prevention’s (CDC) Universal Data Collection (UDC) project. Haemophilia. 2009 Nov;15(6):1281-90. PMID: 19637999.
- Plummer ES, et al. Prominent forehead hematomas (“goose-eggs”) as an initial manifestation of hemophilia. J Pediatr. 2013 Dec;163(6):1781-3. PMID: 23968747.
- Kitchens CS. Occult hemophilia. Johns Hopkins Med J. 1980 Jun;146(6):255-9. PMID: 7382250. PMID: 7382250
- MASAC Document 257 – Guidelines for Emergency Department Management of Individuals with Hemophilia and Other Bleeding Disorders. Published 2019. Link.





1 thought on “emDOCs Podcast – Episode 81: Hemophilia”
That was really very informative.