Author: Shane O’Donnell, DO (EM Resident Physician: UTSW – Dallas, TX); Zachary Aust, MD (Assistant Professor of EM/Attending Physician: UTSW – Dallas, TX) // Reviewed by: Alex Koyfman, MD (@EMHighAK) and Brit Long, MD (@long_brit)
Welcome to EM@3AM, an emDOCs series designed to foster your working knowledge by providing an expedited review of clinical basics. We’ll keep it short, while you keep that EM brain sharp.
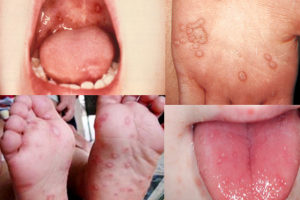
A 23-year-old female with no past medical history presents to the ED for prolonged bleeding from her tooth extraction earlier the same day. Her dentist was planning on removing all her wisdom teeth but stopped after the first extraction due to inability to achieve hemostasis. She has never experienced this kind of bleeding before but notes that recently her gums often bleed when brushing her teeth and describes her last few menstrual cycles as “heavier” than usual. She is not on any blood thinners and was adopted at birth without record of family medical history.
On exam, her vital signs include HR 92, BP 118/78, RR 14, O2: 98% on RA, and 37.1°C. Tooth number 17 appears to have been extracted, and there is blood-soaked cotton balls and gauze between the buccal mucosa and the cavity where tooth 17 used to be. Upon removal of the gauze, you notice a slow oozing of blood from the extraction site. No trismus, sub-mandibular swelling, or other signs of potential airway compromise are present.
What are some of the bleeding disorders on your differential given this clinical presentation?
Answer 1: Von-Willebrand Disease
Answer 2: Hereditary Hemorrhagic Telangiectasia
Answer 3: Immune Thrombocytopenic Purpura
Answer 1: Von-Willebrand Disease (vWD)
Background:
- Most common inherited bleeding disorder
- Disorder of primary hemostasis
- Von-Willebrand factor facilitates the binding of platelets to subendothelium and prolongs the half-life of Factor VIII1,2
- Various Types3
- Type 1 – Most common. Quantitative reduction in Von-Willebrand factor
- Acquired Type – Can occur in conjunction with other illnesses or medications
History:
- Look for common age-specific conditions that may expose a patient’s underlying vWD.
- Infants: Prolonged bleeding after circumcision
- Children: Epistaxis >10 minutes
- Any age: easy bruising, prolonged bleeding with tooth extraction or surgery
- Adult females: Upwards of 60-90% of females with vWD have heavy menses4
- Can be acquired/associated with5:
- Medications (e.g., Warfarin, DOACs, etc.)
- Illness (e.g., Uremia, DIC, etc.)
- These are often termed Acquired Von-Willebrand Syndrome
Physical Exam:
- Assess for bleeding at skin and mucosal sites primarily
- Identify and evaluate for any life-threatening bleeding
- Unexplained bruising should be evaluated
- Consider the possibility of NAT or abuse if the history appears inconsistent
Evaluation:
- ED evaluation should focus on stabilization and referral to a hematologist
- Labs:
- CBC – May not be abnormal
- Thrombocytopenia can be seen in type 2B
- Prolonged, undiagnosed vWD may result in microcytic anemia6
- aPTT / PTT – Can be prolonged due to the role Von-Willebrand factor plays in prolonging the half-life and stabilization of Factor VIII
- Many specific vWB labs that are beyond the scope of ED evaluation but may be helpful for the specialist referral1
- CBC – May not be abnormal
- As always, start with the ABCs and move to specific vWD interventions if the history and clinical picture supports it as the etiology for any hemodynamic instability.
- Desmopressin (DDAVP)7
- IV: 3 mcg/kg (maximum dose, 20 to 30 mcg) in 50 mL saline over 20 minutes
- Nasal Spray: Weight >50 kg: 300 mcg (1 spray in each nostril); weight <50 kg: 150 mcg (1 spray in 1 nostril)
- Tranexamic Acid
- 20-50mg/kg q6-8hr
- Crush tablets into 10-15mL of water, swish/hold in mouth for 5 minutes before swallowing
- Absorbed by the buccal mucosa and secreted into saliva
- Von-Willebrand Concentrates / Recombinant vWF / IVIG / Antifibrinolytic Agents
- Dosing in consultation with hematologist
- 20-50mg/kg q6-8hr
Answer 2: Hereditary Hemorrhagic Telangiectasia (HHT)
Background:
- Formerly known as Osler-Weber-Rendu syndrome
- Vascular disorder with autosomal dominance inheritance
- Prevalence between 1:5000 and 1:80008
- Iron deficiency anemia, GI bleeding, and epistaxis are the most common manifestations9
History:
- Family history will be an important clue but onset of HHT is typically not apparent at birth. Rather, it develops over years
- Focus on history of unexplained episodes of bleeding
- Epistaxis is often the first and most common manifestation with a majority of HHT patients reporting daily nosebleeds9
- Recurrent GI bleeds can occur in up to 1/3 of people with HHT10
- Most commonly from telangiectasias in the stomach > duodenum > colon
Physical Exam:
- Look for evidence of mucocutaneous telangiectasias
- Findings may be subtle in younger patients10
- Lips
- Tongue
- Buccal mucosa
- Fingertips
- Visceral involvement can be challenging to assess for during physical exam
- HHT paradoxically increases the likelihood of VTE and arterial thromboses11,12
- Signs of heart failure may be an important clue, as AVMs can predispose patients to high-output cardiac failure13

Evaluation:
- Visceral involvement of HHT is best characterized by CT and/or MRI imaging
- Genetic testing is low-yield in the ED and should be reserved for outpatient evaluation
- International Consensus Criteria for diagnosis (3+: Definite, 2: Suspected, 1: Unlikely)14
- Spontaneous and recurrent epistaxis
- Multiple mucocutaneous telangiectasias at characteristic sites
- First-degree relative with HHT
- Visceral involvement
Management:
- Therapies will be directed based on the vascular culprit15
- Epistaxis from HHT has many outpatient options that can include monoclonal antibodies but our goal in the emergency department is topical therapies to achieve hemostasis
- Tranexamic acid has been proposed as a second-line therapy, but controversy remains due to the theoretical prothrombic state from HHT12
- GI bleeding often self resolves but may require endoscopic ablation
- Pulmonary AVMs often self-resolve but may require embolization in certain cases
- Hepatic AVM embolization should be avoided due to significant increase in morbidity and mortality
- Epistaxis from HHT has many outpatient options that can include monoclonal antibodies but our goal in the emergency department is topical therapies to achieve hemostasis
Answer 3: Immune Thrombocytopenia (ITP)
Background:
- Also known as Idiopathic Thrombocytopenic Purpura or Immune Thrombocytopenic Purpura
- Pathogenesis is not completely understood but thrombocytopenia from antibody-mediated destructed is the primary hypothesis
- Can occur at any age but peaks in childhood, young adults, and eldery16
- Primary ITP is an isolated response to platelets whereas secondary ITP happens as a result of a larger pathologic process17
History:
- Preceding viral illness has been associated with ITP in children but is not required for the diagnosis
- Mucosal bleeding such as epistaxis or bleeding gums can be an important clue
- Ask about any new findings on the skin
- Secondary ITP will have variable presentations
Physical Exam:
- Skin findings are common, specifically petechiae and purpura
- Purpura should not be palpable, as palpable purpura should raise suspicion for a vasculitis etiology
- Hemorrhagic mucosal bleeding
- To make things more complicated, patients may be asymptomatic and have no physical findings
Evaluation:
- ITP is a diagnosis of exclusion
- A CBC with platelet levels below 100 x 109/L is required for diagnosis16
- A blood smear should be obtained once thrombocytopenia has been identified
- Quantitative immunoglobulins and a direct antiglobulin test should be obtained in both adults and children17
- Adults should also be evaluated for common etiologies of secondary ITP including HIV, Hep B, and Hep C
Management:
- Any life-threatening bleeding should be addressed before consultation with hematologist for any further recommendations17
- Platelet transfusions have demonstrated success in achieving hemostasis17
- Prednisone: 1mg/kg with a max of 80mg
- IVIG: 1mg/kg
- Children:
- No bleeding or mild bruising/petechia can be managed outpatient regardless of platelet level
- Any bleeding with mild bruising/petechia can be addressed with one of the therapies below17
- Prednisolone: 4mg/kg/day with a max of 400mg/d for 4 days
- IVIG: 1mg/kg
- Anti-d Ig: 75mg/kg if Rh+, no anemia, and has a functional spleen
- All patients suspected of ITP should have hematology consult and outpatient referral
- Admission should be considered for any patient that required treatment for bleeding, uncertain diagnosis, or inability to attain hematology follow-up within 72 hours
Pearls:
- vWD is the most common inherited bleeding disorder, but do not forget to evaluate for the acquired form
- DDAVP should be considered for all patients presenting with vWD
- HHT can present at any age but most commonly is associated with recurrent epistaxis
- ITP can be an incidental primary finding or secondary to another disease process
- Platelet transfusion has demonstrated success in both children and adults with the right indications and admission should be considered for any bleeding that required intervention

A 3-year-old girl is brought in after her mother noticed a rash and bruising over her trunk and extremities. She also has intermittent epistaxis over the past few days. She had an upper respiratory illness two weeks ago but otherwise is well. Examination reveals a well-appearing child with scattered petechiae. Hemoglobin is 12 g/dL, WBC 8,000, INR 1.0, and platelets 8,000. Which of the following is the most appropriate initial treatment?
A) Corticosteroids and intravenous immunoglobulin
B) Observation
C) Platelet transfusion
D) Splenectomy
Answer: A
The child has idiopathic thrombocytopenic purpura (ITP), an acquired autoimmune disorder that results in platelet destruction. It often follows a viral illness and is more common in children than adults. It is characterized by thrombocytopenia in the absence of other bone marrow pathology. Signs and symptoms include petechiae, purpura, and gingival bleeding. Management depends on degree of thrombocytopenia and presence of bleeding, and should be performed in consultation with a hematologist. In general, patients with platelet count of 10,000-20,000 µL and mucosal bleeding or those with platelet counts < 10,000 µL and no bleeding are treated with corticosteroids or intravenous immunoglobulin (IVIG) or both. Asymptomatic patients with platelets > 20,000µL can be observed, as the condition is often self-limited.
Observation (B) is indicated for asymptomatic patients with platelets > 20,000/µL. Platelet transfusion (C) is only indicated for life-threatening bleeding after administration of IVIG and steroids. Splenectomy (D) is rarely required for ITP and is only considered in refractory cases.
Further Reading:
- https://wikem.org/wiki/Von_Willebrand_disease
- https://rarediseases.org/rare-diseases/hereditary-hemorrhagic-telangiectasia/
- http://www.emdocs.net/em3am-idiopathic-thrombocytopenic-purpura/
- https://pedemmorsels.com/von-willebrand/
References